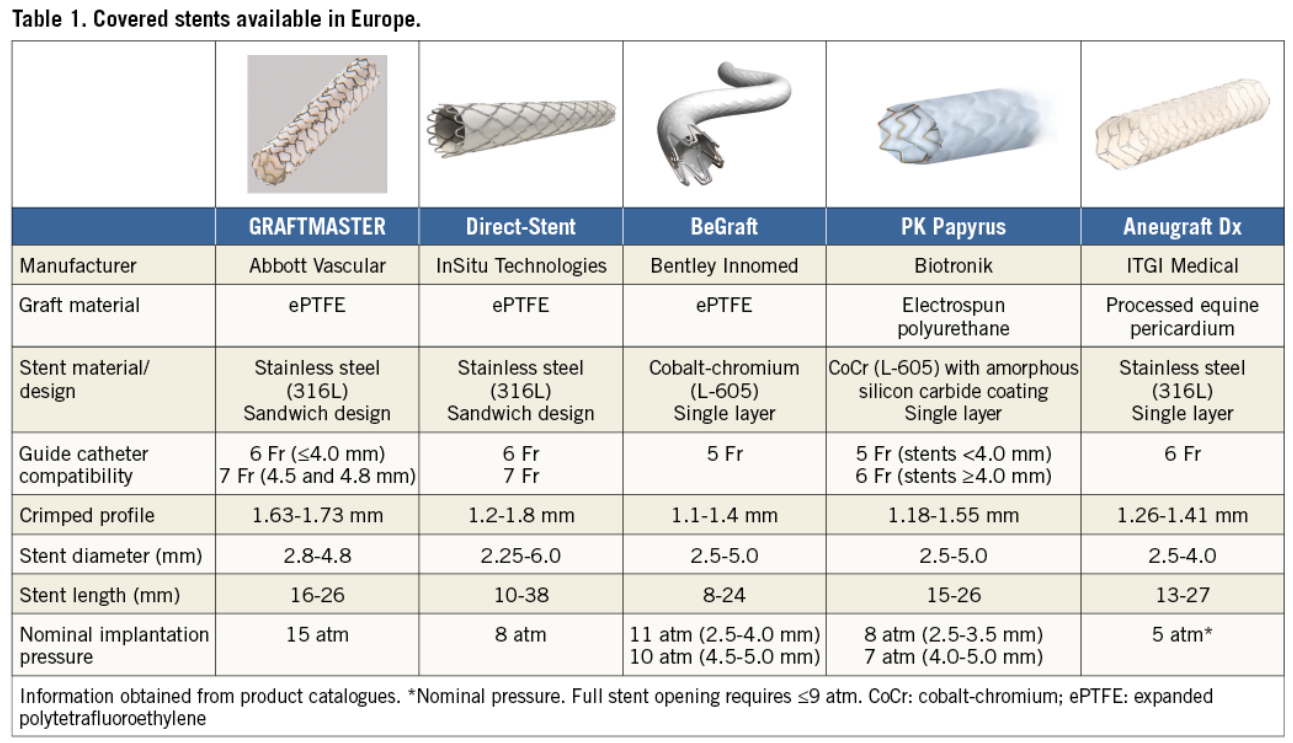
Covered stents are exclusively reserved for coronary artery perforations. Yes, that’s what we think. There has been limited exploration regarding the value of covering the complex lesions, which could prevent future coronary events .
It is possible, covered stents might play a extended role , other than perforations as in complex .friable thin capped lesions . As of June 2025 , haven’t found any such study in cardiology literature.
The recently released PREVENT study argued for PCI for patients with vulnerable high risk plaque. Ironically , it is found plaques with very thin cap ie <50microns are at risk of rupture by the radial stress of struts in the immediate or late follow up.
The thought of this study came when we witnessed high recurrent events, due to plaque prolapse, TCFA injury, new plaque ruptures, micro emboli. no reflow etc in patients with complex lesions.
Any past studies done on this aspect ?
There have been some attempts to use covered stents in degenerated venous grafts. Also, the M-Guard stent system was used in the past to seal thrombus during primary PCI. Both showed mixed results. (Gracida 2015)
Are we ready for a trial with a far fetched Imagination ?
What about jacketing and sandwiching the coronary lumen internally with a synthetic layer of tissue? That can potentially prevent recurring events indefinitely. (It is like making a native coronary artery into a Teflon-coated tube.) The proposal may look crazy until we find a inert layer of synthetic tissue to false roof the coronary lumen. But someone can make a start.
Final message
Covered stents are not just meant to arrest blood leaking outwards, in case of perforation , it can also be used seal high risk plaques, that ruptures and leaks its content into the lumen.
*In the following document, a brief outline and proposal is written about such a study. Whoever wants to do such a study, may use it. I wish I could be an external adviser, as I am no longer attached to a teaching hospital or research center.
Postamble
Before , we begin such a study, one may look at the long term outcome of patients who had already received covered stents for perforations. This is important because, PTFE’s pro-thrombotic potential and need for additional vigilance is yet to be defined.