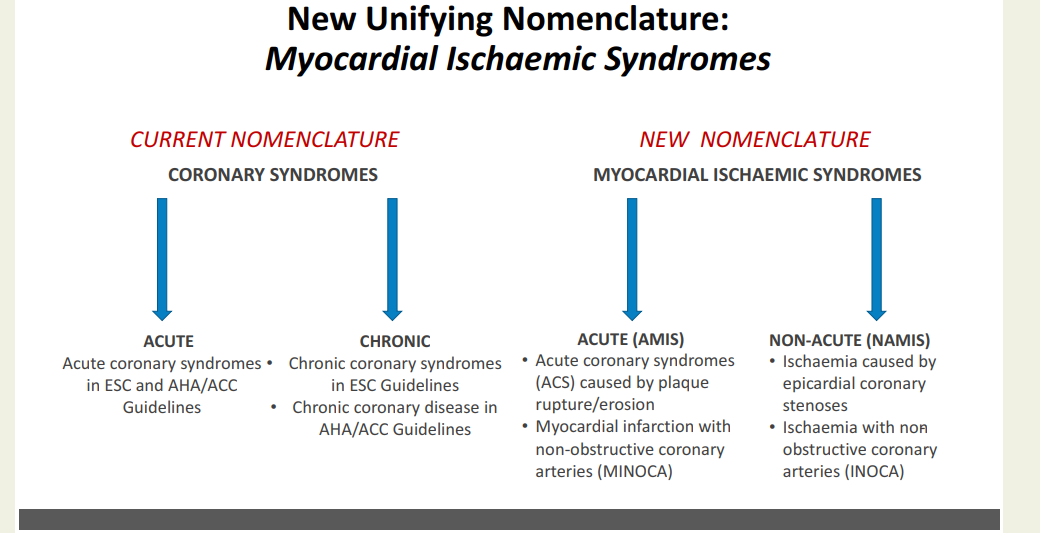
CAD : It is been called IHD, CCD, CAHD, CCS, etc. Now there is a new proposal from European Heart journal (with an Impact factor of 48 ) to change the nomenclature of chronic coronary syndrome to NAMIS : Non acute myocardial Ischemic syndrome (Ref 1)
What is the purpose & need to change the current terminology ?
The proposed AMIS and NAMIS classification shifts the clinical paradigm from coronary anatomy to myocardial function. For decades, terms like “coronary artery disease” overemphasized epicardial stenosis, neglecting the reality that ischemia frequently occurs without structural blockages. Data shows that up to 40% of symptomatic patients lack macroscopic obstructions, suffering instead from microvascular dysfunction, vasospasms, or myocardial bridges. This new term NAMIS can bring in the phenotypes like INOCA and MINOCA into the CCS without any conflict. Further, replacing vague adjectives like “stable” or “chronic” with “non-acute” reflects the high long-term residual risk of cardiac events. I also think, this new framework is meant to unify the European and American guidelines, where they use differing terminologies.
Counter Point : Do we really need this ?
Purpose of any new classification is , it should have a clear impact on patient management. But, we often find, scientific committees periodically take pride in changing the nomenclature akin to make a academic fashion statement .Within a few short years, we migrated from stable CAD to SIHD, switched to CCS , CCD etc . The fact is , for the patients it is the same angina for which they seek relief. They simply don’t bother whether doctor label their condition a syndrome or disease .
It is an irony, we paint the lipids as chief villains in the genesis of atherosclerosis and CAD. The fact of the matter is, it is the protein component that hides among the lipid jelly, with multiple folds and stinging tails, that ultimately help the LDL to penetrate the intact endothelium . These are called , apo-lipoprtiens. The fate of lipid molecules is determined by these apparently humble looking proteins .It must also be noted the even the goodness of HDL vanish if its apo-liprotein fails to co-operate . The Apo proteins are labeled differently in each sub fraction of lipids. Its Apo A 1 in HDL Apo B 100, in LDL and Apo C in VLDL.
Playing with English Alphabet A
Scientists can be casual , though not intentional when naming newly detected molecules. The major nomenclature issues are with Apo A , Apo a , Lip a These three entirely separate proteins with nearly identical names, should not be confused with. A subtle shift between a lowercase letter and a capital letter completely changes the meaning, taking us from a highly dangerous cardiovascular risk factor to a healthy, protective one.
Apolipoprotein A : This is a highly beneficial protein that defines the HDL function.(Ofcourse one must remember too much of HDL can be toxic as well .In fact when it exceeds 60 mg ,it will become dysfunctional, rather harmful.This is the U curve phenomenon of HDL)
Apolipoprotein(a) : This is the sticky, genetically fixed protein tail found only on Lipoprotein(a). It promotes blood clotting and triggers arterial inflammation.
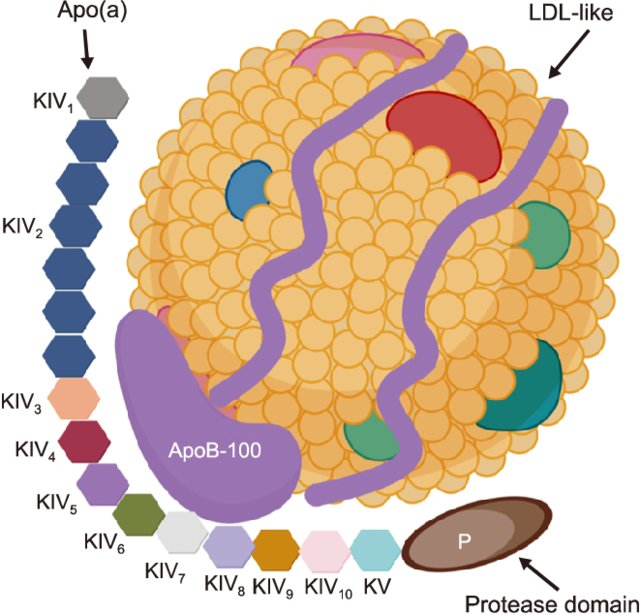
Lp(a) : It is formed when a single Apo(a) protein tail binds to the Apolipoprotein B-100 (ApoB) wrapper of a standard LDL like particle. Because of this hybrid structure, Lp(a) possesses dual pathogenic properties. LDL core delivers cholesterol into the arterial wall, while the attached Apo(a) tail promotes localized inflammation and thrombosis . Lp(a) plasma levels are strictly determined by inheritance and remain highly stable throughout an individual’s life.
Lipoprotein(a) structure. Lp(a) is composed of two parts: one part is low-density lipoprotein (LDL)-like particles with apolipoprotein B100 (apo-B100), and the other part is the stinging tail apolipoprotein a [apo(a)]
Among the above three molecules, lipoprotein(a) is the celebrated one kept in the limelight. It is strongly determined by genetics and is a predictor; rather, we can call it a marker of LDL virility (like Apo B 100). It is entirely produced in the liver, its level is claimed to be static lifelong. Obviously, it cannot be true, as it would heavily dependent on liver function. Further, different vendors have come into the market with this, and they have different normal ranges.
Final message
When reading the lipid molecules, please focus on the English . A capital A vs. small a makes a huge difference between harming and healing the patient. Of course, we must be thankful, as of now, we have don’t have no small b or small c Apolipoproteins in lipidology.
India is sort of burning, with students on the streets for exam reforms.The problem started with the leakage of question paper for National Entrance and eligibility test for medical college( NEET) Government is on the back foot. The prime minster has come out, assured the students and their concern will be taken care of. Yes, it is true greedy cartels are exploiting the system. But, we are ignoring a big hidden moral failure in the whole issue.
However, It is very convenient to march through the streets pointing fingers at the state, but blaming the government alone is not enough to fix a rotting system. We love to demand absolute structural perfection from institutions while completely ignoring the mirror of reality. It is us, the public that fuel and maintain the corruption supply chain through a collective, desperate willingness to bypass merit at any cost.
Let’s be honest, even a un-breachable testing infrastructure cannot save an education system where parents and students are actively looking for illegal shortcuts. When families willingly treat leaked question papers as a premium commodity, they are not victims they are the core investors funding the black market. I wished, I could find at-least one banner conveying this fact too.Unfortunately, it can never happen.
True reform cannot happen through government orders alone. It requires a brutal confrontation with our own societal hypocrisy and a complete dismantling of the demand side of corruption.
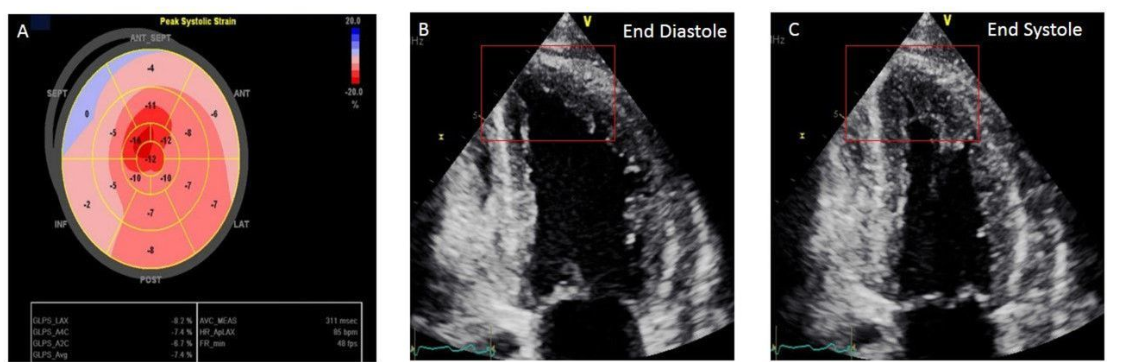
TAPSE the most celebrated RV function index misses significant Right Ventricular (RV) dysfunction in an estimated 30 to 50% of patients. ( when relying solely on it).It fails to detect underlying impairment because it only measures the longitudinal shortening of the RV base, largely ignoring global chamber contraction.
Mechanism attributed to the unreliability of TAPSE
Ventricular Interdependence: The RV motion is influenced by left ventricular function by the shared IVS contraction.
Chronic Volume or Pressure Overload: Conditions like severe tricuspid regurgitation or pulmonary hypertension cause the RV to alter its contraction geometry. This leads to “pseudo-normalization, of TAPSE” it remains >1.7 cm despite dysfunction.
Post-Cardiac Surgery: In patients who have undergone procedures involving pericardiotomy, TAPSE often drops drastically as post pericardiotomy the RV radial function exccedds longitudinal
Loading Dependency : TAPSE is highly influenced by preload and afterload conditions.
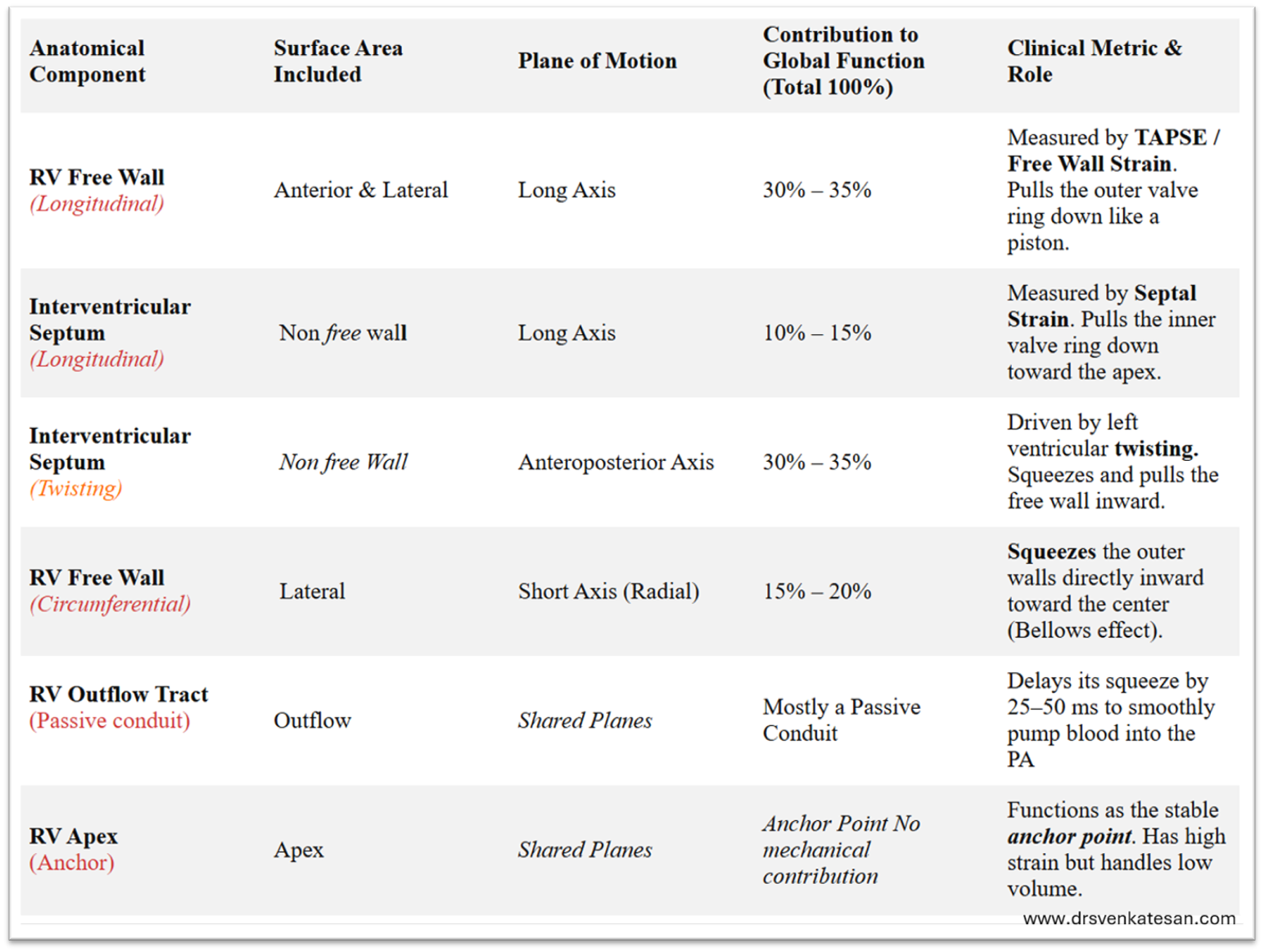
The following table summarises the various components and it’s contribution to RV contractility.
*Contrary to the popular belief, there is overlap between free wall contraction and longitudinal contraction. In fact, there is no clear definition for RV free wall. Logic tells us, any part of RV which is not formed by IVS can be considered free.
*Importantly, Longitudinal contraction has a two components free wall as well as a septal axis. TAPSE measures only lateral or free wall component of longitudinal function. It is less influenced by septal long axis function.
*The long axis function of RV is influenced indirectly by the LV function also as both AV valves are attached to same ring.
*If we want to assess pure RV function the best index is RV free wall circumferential or radial contraction or strain.
*In various clinical situations like PH or acute pulmonary embolism the pressure distribution is non-uniform making the assessment of RV function difficult. Fractional shortening of area is a fair index.RV wall motion abnormality can be subtle yet a serious marker of RV dysfunction.
Final message
RV function assessment is complex and often incomplete. The habit of relying only on TAPSE, is not a high quality scientific practice.
Postamble : Apart from the contractile function, we don’t know at what RV pressure RV begins to dilate. This is different in acute vs chronic elevation. We also don’t know which patient will show RV hypertrophy, and which group prefers dilation. May be, all these are academic and has little significance at the bedside. However understanding this is essential to assess the response to RV inotropes and newer RV assist devices.
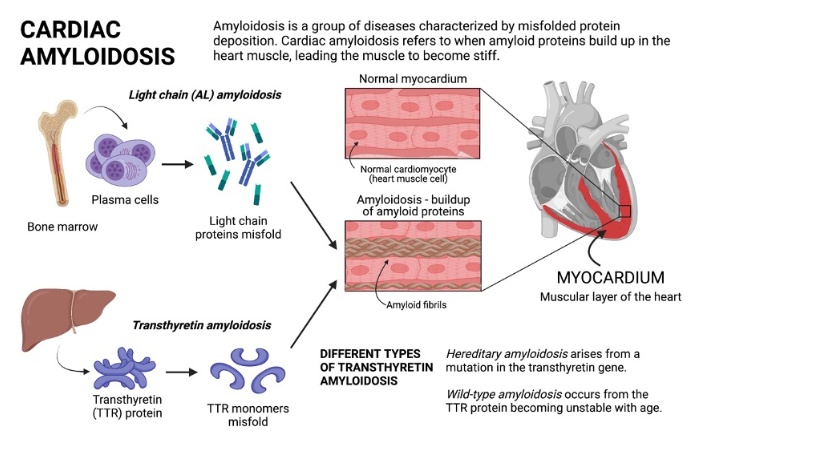
Amyloidosis is a proteostatic disease , meaning a progressive disorder of extracellular deposition of insoluble protein fibrils in tissues.It affects , brain, kidney, nerves and almost any organ.Crucially, amyloid is an interstitial disease, accumulating strictly in the spaces between cells rather than inside them. This interstitial buildup exerts physical pressure, alters tissue architecture, and finally invading the cell and hence the organ function.
The primary culprit is a normal transport protein transthyretin (TTR) (same as prealbumin) It is synthesized in the liver as a stable, four-part tetramer structure that safely circulates in the blood. Under stressful or aging conditions, this tetramer destabilizes unstable monomer pieces. These monomers undergo misfolding, polymerizing into rigid cross-beta-sheet amyloid fibrils. These abnormal ATTR fibrils escape cellular degradation.
Cardiac amyloidosis
Cardiac amyloidosis , presents as mainly as restrictive cardiomyopathy, but can be seen in any chronic heart failure associated with LVH, especially in Aortic stenosis, where it can involve both the valve and myocardium.There are multiple reasons why we diagnose amyloidosis more often than before. But , one thing is sure, it is not due to increasing incidence in the population , but more of a high awareness and availability of advanced cardiac imaging .
Image source : Agha AM,. Role of cardiovascular imaging for the diagnosis and prognosis of cardiac amyloidosis. Open Heart. 2018;5:
What is the confirmatory test for Amyloidosis ?
It was classically diagnosed by tissue biopsy with Congo red staining, showing “apple-green” birefringence under polarized light microscope. Mass Spectrometry, Immunofluorescence is then used to decode the protein composition. Currently , with typical patterns of cardiac imaging we can diagnose with confidence without biopsy. While echocardiography is good enough , MRI and technetium-99m pyrophosphate scans are crucial.
What are the specific treatment option for amyloidosis ?
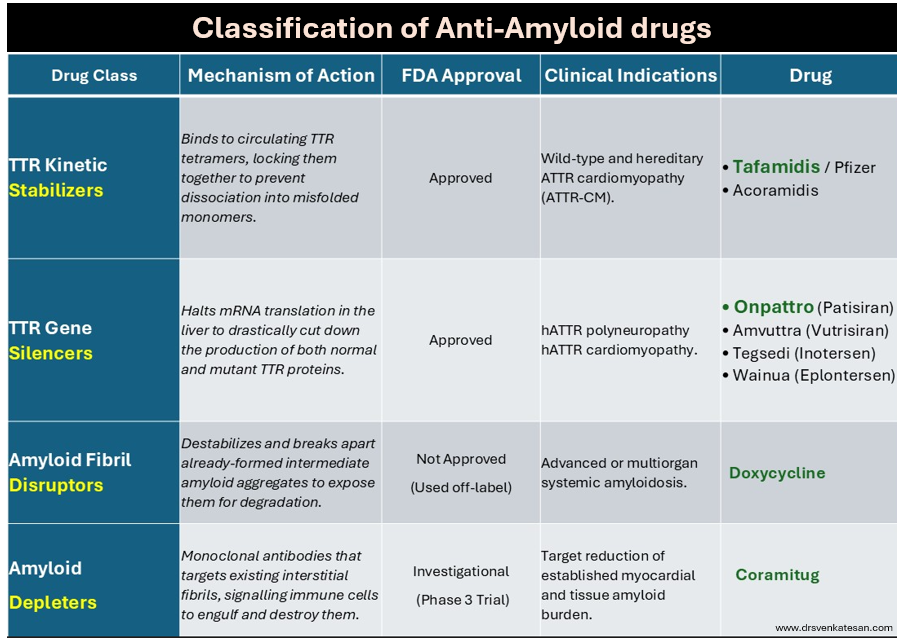
Until recently, Amyloidosis, has been considered as irreversible degenerative disease .Now we have strategies to reduce progression or even regress the Amyloid deposition. Many drugs are still being studied with varying success.
Following table summarise the different pharmacological modalities to tackle Amyloidosis.
Final message
A limitation of currently approved anti-amyloid medication such as Tafamidis or Onpattro , is that it cannot be reversed and only be prevented.Emerging technologies like monoclonal antibody depleters hold promise to actively clear out existing deposits, potentially turning it to a truly reversible condition. Chronic fibrotic pathways from triggered fibroblasts are closely linked to Amyloidosis. Hence, antifibrotic drug research should go hand in hand with Amyloidosis.
Pulmonary embolism continues to be an intriguing entity, both in the diagnosis and management. The spectrum is wide, from a clinically insignificant, or totally unrecognized episode to a massive cardiopulmonary event causing sudden death. Similarly, treatment can either be conservative (even sending the patient home ) or lead to aggressive cath lab intervention.
The task of PE risk stratification
Obviously, risk stratifying is the mainstay in the management.The fact that we are grappling with multiple risk stratifying scores like PESI, simplified PESI or HESTIA expose our limitations. To segregate , low and high‑risk categories do not demand much expertise. It is the intermediate‑risk group that plays havoc in the ER. It is based on RV function and biomarkers like troponin and D‑dimer. But it was soon realised clinical parameters like blood pressure and hemodynamic stability are the more powerful markers of risk.
However,the reality is, many patients in the intermediate‑risk group are rushed to the cath lab, either due to overestimation of risk or fear of potential escalation to high risk. A third reason,is often non‑academic, it’s done for showing our expertise.
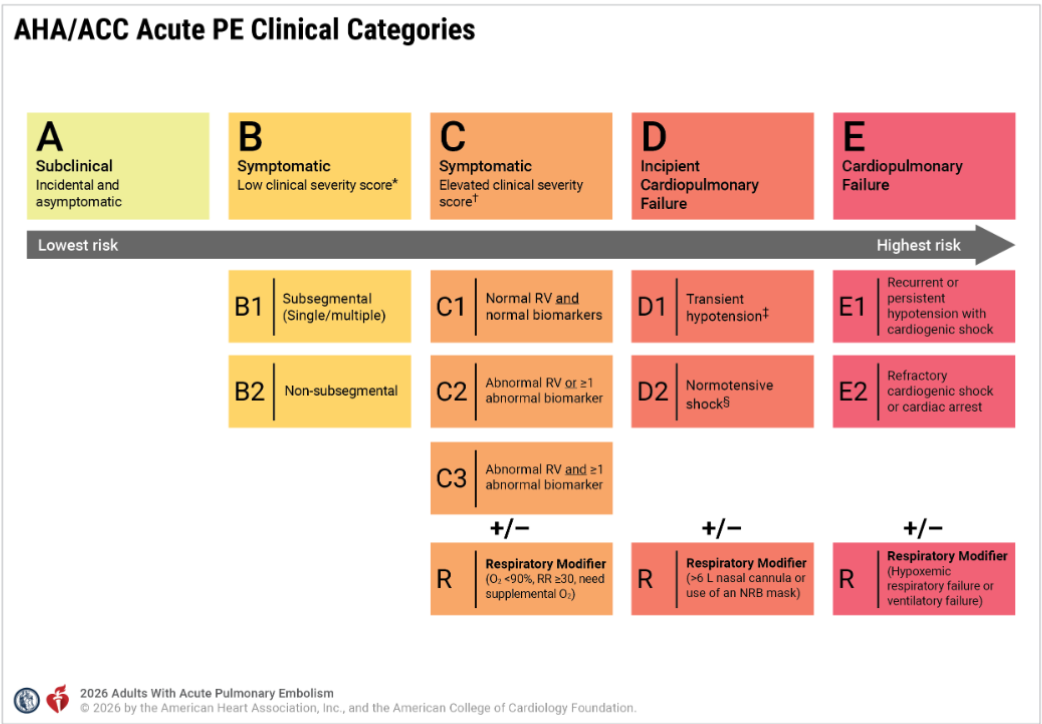
Now, to solve the puzzle of intermediate risk, we have created two new subcategories, C and D. ACC/AHA has been very careful in defining and labeling the entities. Still, there are a lot of gaps that need to be fixed.
In this post let us confabulate few aspects of the new grading of PE and in particular the curious case of D 2 shock .
series of iFAQ ( infrequently) asked questions will from the basis of this discussion
Does category C to D progress in a sequentially worsening hierarchy?
Though it looks that way, the clinical presentation rarely follows a sequential order.
Why is BP is not given adequate weightage in Category C?
A normal blood pressure is a mandatory baseline requirement to enter Category C. However, BP is an important factor used in the clinical severity assessment (PESI etc) .It is not clear why , the committee did not explicitly use the term Category C with reference to BP.
Why is the word “symptomatic” missing in Category D?
The word “symptomatic” is deliberately omitted from the title and subcategories of Category D in the 2026 AHA/ACC Pulmonary Embolism guidelines because symptoms alone are no longer a differentiating factor at these higher levels of severity. Alos, the patient needs to be alert, to tell the symptoms.
Does that mean symptomatic Class C is less risky than asymptomatic Class D?
Yes. A symptomatic patient in Category C is at a significantly lower risk of dying or collapsing than a patient in Category D.A patient in Category C might feel worse because they are aware of their chest pain. A patient in Category D may not complain of pain at all, perhaps because they are in altered sensorium.
What is D2 shock, and when and how do we diagnose it?
First, the patient must fulfill the baseline criteria of Category C, which includes right ventricular dysfunction and positive biochemical parameters of either Troponin or NT-Pro BNP. Then, they must have at least one parameter to confirm shock. Under Category D, the term normotensive shock defines the D2 category, where the blood pressure is normal.
The signs of shock include:
Lactate greater than 2 mmol/L.
Evidence of acute kidney injury, meaning urine output is less than 0.5 mL/kg/hr.
Altered mental status changes.
A cardiac index less than 2.2 L/min/m2.
A mean arterial pressure less than 60 mmHg.(In D2 shock the MAP more than 60 mmhg)
Is elevated lactate mandatory to diagnose D2?
No. Any single criteria from the above list is enough. Oliguria alone or even altered mental status is sufficient.
Can D2 shock be diagnosed based only on altered sensorium?
Yes. it is possible to diagnose Category D2 based only on a vague, mild, or temporary change in mental status .This carries a huge risk of falsely classifying a stable Category C patient into the more critical Category D. To prevent this error, the 2026 AHA/ACC guidelines has strict definition of Category D2, ie the altered sensorium must be profound and persistent.
Why Category D1 patients can show a fall in BP, while Category D2 has normal BP?
In PE, the right heart is overwhelmed by the acute rise in RV afterload, causing a short term drop in the volume of blood until the RV recovers. This is the mechanism of fall in BP D1.In Category D2, the blood pressure stays completely normal (90 mmHg or higher), but the patient may be in ongoing organ shock spiral. In D2 the RV is failing just like in D1, but the patient’s sympathetic response fires at it’s peak with maximum vasoconstriction to sustain the BP and life.
Why D2 shock sounds a mystery ?
It may look implausible that a patient in D1 can have low blood pressure, but when they worsen, they seemingly gain blood pressure to reach D2 and get the tag of normotensive shock. In the bed side this is not true. The hidden clinical reality is, D1 and D2 are not meant to be viewed as sequential, worsening hierarchical stages. A patient can move from Category C to D1 or D2 directly, and a direct jump from Category C to Category E is also possible without spending any time in Category D.
Dynamics of the C-D-E categories
It becomes problematic to have the term normotension in the title itself in D2 . This implies ,a patient with hypotension and underperfusion should exit from D2 shock. This could mean a worsening patient paradoxically enters Category D1 or Category C or E. To be precise, a fall in blood pressure represents is really a sign of worsening or not is debatable and decided on patient to patient basis. Also both category C will also feature a normal blood pressure. This can be mixed up with D2 if organ perfusion markers like oliguria occur transiently. This trichotomy between hemodynamic stability, organ perfusion , versus respiratory stability needs to be addressed. The PERT team need to be vigilant on this asseement.
Do we need to sub classify D2 shock ?
In realistic observations, the most common cause of normal BP in D2 shock is the intensive treatment these patients receive with RV inotropes and systemic catecholamines. Perhaps we may need to add another rider to Category D2, such as Normotensive with drug support versus normotensive without inotrope support. This would help us know the exact direction in which these patients will progress.
Can Category C3 be riskier than D2?
This is tricky and indeed seems possible . Category C3 can exceed the morbidity of Category D, especially when it features the respiratory modifier as C3R. It must be recalled that both the C and D categories present with right ventricular dysfunction and positive cardiac biomarkers, but their primary organ failure differs. A Category C3R patient experiences an acute oxygenation crisis. Conversely, a Category D2 patient, even with normal lactate, suffers from occult circulatory shock (an adequately functioning liver can clear the lactate efficiently.) Though C3R appears more dramatic at the bedside due to the need of respiratory support , Category D2 carries a higher mortality.
Final message
“It looks like treating patients with PE is much easier task than risk stratifying them“
Pulmonary embolism management is a fierce fight between the right ventricle, the occluded pulmonary arteries and the lungs.The first 48 hrs is crucial. RV shock incidence rapidly reduce after this time window. We realised very late, the total thrombus burden actually has a poor correlation with the final outcome. Most episodes of PE can be managed medically.
Balancing pharmacological lysis versus catheter-based lysis requires a very delicate skill. While most patients are eligible for standard lysis, only a very few truly benefit from catheter-based therapies. The rate of RV failure and mortality drops dramatically once the right ventricle rises to the challenge. Therefore, there is no clinical point in doing a catheter-based lysis if the blood pressure is normal and the right ventricle is already recovering.
Finally and most importantly, the alarming entity D 2 (Normotensive shock) should not cause unnecessary panic. If the, saturation and RV function remains good or shows signs of recovery , catheter based modalities can be avoided.
“Medical care is sought after and delivered with all sincerity and kindness. It is never meant to be sold over the counter” My mentor and respectful professor used to tell us, many decades ago during our undergraduation days. That was a bygone era. Forget it, let us come to the real world.
This ad came two days ago in a India’s leading daily :The Hindu
With all three limbs of the medical profession, namely patient care, teaching, and medical research, under the clutches of commerce and industry, there is no surprise at all, to find this ad in the mainstream media.
How big is the AF burden ?
Atrial fibrillation is a common arrhythmia of the elderly. In that sense, it is more prevalent in the western population where life expectancy is high. AF is more of a nuisance arrhythmia in an otherwise normal heart, which requires stroke prevention strategies like antiplatelet agents or OAC/DOACs. In a hemodynamic perspective it is largely benign arrhythmia , 95% of which can be managed with simple rate modulating drugs like Digoxin and Beta blockers .The costly and often riskier catheter based rhythm restoring modalities still have questionable value except in a fraction of the AF population.
What could be the aim and intention of the above advertisement?
Big companies want to sell complex medical devices and procedures for AF directly to the public.The funny thing in the ad is , while India is grappling with the task of providing basic health amenities like ECG machines in every primary health center , yet this ad expresses serious concern about the low penetration of a new, cost-ineffective, technology called “pulse field ablation” to treat AF.
Final message
I think, Govt of India should seriously consider banning such adverse health Influencing ads. direct to public.
The fact that regurgitant lesions are well tolerated in pregnancy, by no -way means, women with stenotic lesions always fare badly. Valves do have reserve excess orifice. This means, it can handle the increased blood volume of pregnancy . A stenotic valve do elevate its gradient from the baseline. This increase in gradient is essentially due to augmented cardiac output and not reduction of orifice size.These elevated gradients sustain the required stroke volume and cardiac index, throughout the pregnancy.
Aortic stenosis in pregnancy
Since , most pregnant women are young, the LV function is good enough to sustain the high gradient. It is also possible, the increase in gradient can be spurious , as it is more to do with Doppler mathematics, as we convert velocity to pressure with a simplified Bernoulli equation.What we really bother about is the stroke volume
The folowing table gives a rough course of Aortic stenosis in pregnancy.
Pregnant women* with heart disease has to cross at-least three hemodynamic hot spots during the tenure. The first task comes around 20 to 24 weeks. The hemodynamic stress almost reaches 80% of maximum. If the mother doesn’t worsen at the end of 24 weeks, it is very likely she will pass through the rest of pregnancy. Another less stressful milestone is around 32 weeks where , she reaches the peak hemodynamic stress. Not to forget, the most critical period (48 hrs) is in the immediate postpartum, where the stress of labor and uterine involution infuses more than 500 ml into the maternal circulation. (*It is not clear whether the blood loss associated with either normal or cesarean section will negate the stress of volume overload. In fact, there is no study that has specifically addressed this issue in heart disease complicating pregnancy. However , PPH always harm the mother.)
Final message
Somehow, we are more obsessed with gradients than what really matters , ie the stroke volume and cardiac output. The fear of high mortality with increasing gradients is more of imaginary. In fact, it tells us about the reserve LV power.Most of the mild to moderate AS is well tolerated throughout pregnancy. Of course, severe AS requires intensive monitoring or a temporary balloon dilatation as we do pre-TAVI procedure. (Or full-fledged TAVI may be considered as a last resort.)
Ironically, in the current hyper-academic environment, more than the true hemodynamic stress of the mother, the Obstetrician’s mental stress is much, much higher when confronted with any heart disease with pregnancy. There are many untold stories, where obstetricians, (Influenced by of new-age cardiologists) are compelled to pursue risky interventions in pregnancy to fulfill protocols & guidelines.
What happens to trans mitral gradient during pregnancy in mitral stenosis ?
Natural history of gradients in mitral stenosis in pregnancy.
Unlike AS, where the most powerful cardiac chamber of the heart , ie LV is challenged, in mitral stenosis LA has to fight its battle alone, or with help of RV. The risk of acute pulmonary edema is many fold fold high in MS. In countries with rampant RHD, and severe MS, mortality can be high. Still, very selective use of PTMC is recommended in pregnancy even in severe MS .There are numerous case reports of mother and baby crossing the finish line successfully , with the support of experienced obstetrical team (Of course ,this will be called as more of luck though !) The reality is, the professional guilt & fear of not doing a PTMC, often exceeds our confidence, on the resilience and endurance of a young mother’s, compromised heart.
C.May be , an Important link . (Will wait for evidence)
D.No link at all.
Answer:
There has been many postulations linking the two. 1.Cross over of vasoactive substances like serotonin . 2.Paradoxical microemboli that trigger a cortical migraine like aura .3.Transient hypoxemia due to right to left shunt during valsalva strain.
However ,the truth appears to be, it is more of an association , since both entities are very common in the general population.(Up to 20%) .If you ask for a pragmatic answer, it will be determined by the tempting desire to close an innocuous hole in the heart.
Postamble
While, the connection between migraine and PFO is speculative , but its link to cryptogenic stroke may be little more firm, although fear mongering is not warranted . (RoPE criteria: Risk of Paradoxical Embolism)
Answer: You should have guessed it right. If not, read the following, might help you out.
The first and foremost point is to understand the difference between LV dysfunction and failure. LV failure is a clinical diagnosis. LV dysfunction is an echocardiographic diagnosis.BNP can increase due to any mechanical stretch of LV and may rise even with Atrial stretch . Every patient with baseline LV dysfunction will show mild elevation. Sudden spikes can come with clinical worsening. However, to our surprise the NT pro BNP levels is not found to be correlating with severity dyspnea or orthopnea .
Most importantly NT Pro BNP can elevate in isolated subclinical diastolic dysfunction. This makes interpreting its level more complex.
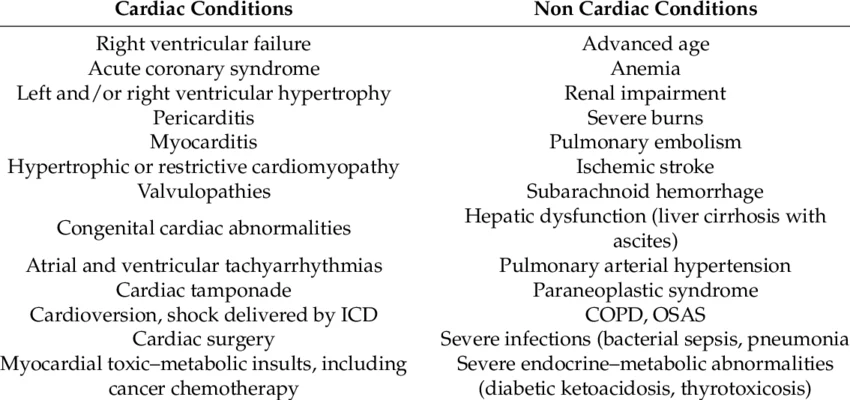
*Additionally, BNP levels can be falsely elevated in renal failure and may be spuriously normal in obese individuals with left ventricular dysfunction. These disparities are due to variations in contributions from preformed (Granular cell depots) and freshly synthesised NT Pro BNP. The later process is severely impaired in obesity.
The following chart illustrates conditions that can cause elevated NT Pro-BNP levels beyond heart failure.
The answer to the question : Every response A to D is correct.
Final message
Relying too much on biochemistry, such as NT pro BNP to diagnose heart failure clouds our clinical judgment.Please realise ,NT pro BNP is not a creatinine equivalent for renal failure .Diagnosis of cardiac failure should focus on clinical criteria and symptoms, with NT pro BNP serving more as a prognostic rather than a diagnostic tool.(The good old Framingham criteria still stands strong & relevant )
The contents of the this blog is being published as Kindle E book , as per the request of many of the readers. Every article will continue to be open source in this site. Again I shall reiterate the book format is not aimed at any commercial intent. It is only to facilitate learning in a single book format Here is the link to book https://amzn.in/d/euhL5vu
Click below to see who is watching this website live !
This site will never aim for profit. Still ,this donation link is added at the request of few visitors who wanted to contribute and of-course that will help make it sustainable .
Please Note
The author acknowledges all the queries posted by the readers and wishes to answer them .Due to logistic reasons only few could be responded. Inconvenience caused is regretted.