
Posted in Uncategorized | Tagged bmj, common sense in medicine, ebm, ethics in medicine, evidence based medicine, experince based medicine, false evidence, jacc, jama network, lancet, medical education, nejm, pitflls of ebm |
The National Eligibility-cum-Entrance Test (NEET-UG) was introduced with the promise of ushering in fairness, standardization, and meritocracy in medical education across India. At first glance, it appears to have succeeded — replacing state-level chaos with a national-level uniform entrance system. However, a deeper analysis reveals that NEET has also created a more insidious structure of exclusion and privilege, particularly through its poorly understood and easily manipulated 50th percentile eligibility criterion. This mechanism has inadvertently (or perhaps intentionally) legalized the backdoor entry of underqualified but wealthy candidates into private medical colleges, all while marginalizing thousands of meritorious but economically disadvantaged students. What we are witnessing is not an accident of policy but a systemic betrayal of the very values NEET claims to uphold.
The core issue lies in the use of the 50th percentile as the qualifying benchmark, rather than a fixed percentage of marks. In a pool of over 12 lakh test-takers, this means nearly 6 lakh students qualify each year, while India has only about 1 lakh MBBS seats — half of which are in government colleges. This creates a vast pool of surplus eligible candidates.
One may argue that such a buffer ensures inclusivity and provides opportunities for students to enter allied medical fields. But in reality, this bloated qualification base primarily serves a far more cynical purpose to feed the commercial engine of private and deemed universities. These institutions, often owned by political syndicates or business conglomerates, need legally “eligible” students to fill their overpriced seats. Thus, NEET’s percentile-based qualification becomes the legal stamp that converts a failing candidate into a paying customer.
The biggest beneficiaries of this system are clearly the private medical colleges. TheY exploit the legitimacy provided by NEET qualification to offer seats to candidates with ranks as low as 5.5 lakhs This is not an aberration; this is the business model. The eligibility net has been cast so wide that even students performing in the bottom 10% are now a valuable market segment for these institutions.
Politicians too have their share in this ecosystem. Many private medical colleges are directly or indirectly operated by political trusts. These institutions flourish under regulatory blind spots and benefit from policies that expand eligibility but do little to control quality. The illusion of merit-based admission helps them deflect criticism while quietly preserving a robust revenue stream. Middlemen, agents, and education consultants also thrive in this system, legally brokering “management quota” admissions for candidates who would otherwise never see the inside of a medical college based on academic merit alone.
The tragedy is not just in who gets in, but in who is left out. Consider the student ranked 45000, who misses a government seat by a few ranks and cannot afford the exorbitant private fees. Meanwhile, a far less qualified peer with is ranked beyond 5 lakhs buys entry into the same profession. The idea that NEET ensures equal opportunity collapses in the face of such economic discrimination.
This skewed dynamic does not merely harm students .It damages the very fabric of the medical profession. The country is gradually producing doctors who may not have entered the system based on ability or passion, but because they could afford to. This threatens the ethical, academic, and clinical integrity of the profession. Over time, it will erode public trust in doctors and healthcare itself. Furthermore, the use of wealth to bypass academic rigor is fundamentally anti-Constitutional. Reservation in education is meant to uplift the socially backward, not to empower the economically elite. By allowing rich mediocrity to flourish, the NEET system insults both merit and social justice.
The system’s design is cunning in its illusion of fairness. NEET’s structure with percentile-based eligibility, decentralized counseling, and layered quotas appears technical and neutral. But it’s a carefully crafted mirage. The Supreme Court rulings that upheld NEET (TMA Pai Foundation vs. State of Karnataka, 2002; and Christian Medical College vs. Union of India, 2020) emphasized fairness and uniformity. Yet today, the same system legally validates an admission model where the top 10% merit students compete for government seats, and the bottom 40% enter through payment, wrapped in a veneer of legitimacy.
NEET should be made as entrance test not eligiblity test
It is time to end this farce. The first reform must be simple but fundamental: eligibility should match seat capacity as UPSC,IAS exams. Only students up to 1.2 times the total number of MBBS seats should be deemed qualified. This ensures that only competent and competitive candidates enter the counseling process.
If India truly wants to select its doctors based on their wisdom and dedication, and not the power of the bank accounts, it must rethink NEET’s qualification model. Otherwise, the country is heading toward a future where healthcare is not just privatized but intellectually bankrupt.
Final message
NEET as model for national entrance is welcome But, only the methodology is wrong .
Realise , when some one says they cracked NEET, by merit, it may sound as if they conquered mount Everest.
Do you know , what exactly it meant this year? All that is required, is to score atleast 144 out of 720 , ie 19% marks, a score, even the back bencher will be ashamed to tell.
It is strange , only medical profession suffers from this. Can you ever think of buying an IIT, Charted accountant or IAS seats , by making lakhs of students eligible through percentile system of examination ?
References
- National Medical Commission (NMC) Regulations on Graduate Medical Education (2023).
- Supreme Court of India. Christian Medical College vs. Union of India, 2020.
- Medical Council of India data on MBBS seats: www.nmc.org.in
- Times of India (2023): “70% of private medical college seats filled by students with ranks >4 lakh”.
- The Hindu (2021): “NEET: Fairness or False Hope?”
Posted in Uncategorized | Tagged neet exam |
Current generation doctors are gifted, can be immensely proud to practice medicine with cutting-edge technologies and advanced medical therapies .Today is the official doctors day in India, in honoring one of most great physician of our times Dr.B.C.Roy on his birth day.
Who celebrates Doctors’ day and for what ?
Sharing here , one of the deeply reflective article about, reality of being a doctor today. Published in today’s Hindu, (July1 2025)India’s National New paper, Opinion Column by Dr C. Aarvinda . It is a 6 minute read, must for all those who truly love our profession.

Courtesy : Dr.C.Aravinda MD, Assistant Professor of community medicine. Thanjavur medical college. Tamil Nadu .India.
While the profession is glorified at every level, there seems to be little to celebrate at a personal level. The same public who celebrate doctor’s day , become a mute witnesses to innumerable attacks unleashed on them on a day-to-day basis across the country. It is a sad truth the medical profession has been hijacked, far away from its original intended destination by both visible and Invisible forces. Many honest, hardworking, and humble doctors are compelled to traverse a turbulent moral landscape.
Final message
Doctors’ day celebration is meant for whom? Realistically, it is the occasion for patients who respect and show love to their healers. The whole idea got distorted in recent times. Definitely, it is not meant for pride hunting and forcible Intrusion by the industry , into the noble profession.
Posted in bio ethics, Ethics in Medicine | Tagged doctors day |
Right now, I am sitting at a yet another national conference on Interventional Cardiology. Two very popular cardiologists, from elite institutes of India are debating on a 60-feet long digital dais, with a flashy background comparable to the Macau skyline. Watching it, are about 600 prosperous delegates , brought from various parts of India. The debate is about, whether to use a single stent or upfront two stent strategy for left main bifurcation disease. The arguments were all too familiar, I just couldn’t concentrate.

I am sure every one will agree ,this topic is being debated for nearly two decades. The answer, we got is crystal clear. 90 % of BFL need just provisional single stent. Rest may require two stents upfront. The quality of the procedure matters more than the technique. Not even Imaging matters much. Of course, we are free to choose DK or various other forms of crush as we like. That’s it. May be, It’s time to we close the shutters on the exclusive and glamorous bifurcation clubs and move on. (until a real Innovation in dedicated bi-furcation stent happens)
The following add on was not part of the debate
*Before any BFL PCI, spend a few silent moments, while the patient is being laid on the cath table . Whether the patient is truly symptomatic, whether he could be a candidate for simple medical management or his lesions are complex enough to deserve a CABG.
Final message
Beginning to wonder, is there a fundamental problem with the current mode of knowledge flow and consumption in the field of cardiology. Why do we keep plagiarizing the same old content in the conferences year after year in spite of being fully aware of the futility? This raises a fundamental issue. As we learn more & more, is there a risk of our wisdom curve getting blunted?
Posted in Uncategorized | Tagged acc esc scai guidelines, bifurcation club, bifurcation lesions, distal vs proximal strut crossing, medical managment for bifurctation lesion, provisionalsingle stent strategy, single or twostent strategy in bfl |
LIMA to LAD anastomotic site lesion ,is an important subset of CAD that can occur either as acute post operative event or an ACS , CCS. Interventional cardiologists have, thus far, been reluctant to intervene in this type of lesion, often refer to a surgeon instead.
Here is case report by by Tahir et al. wherein a 75‑year‑old post‑CABG patient who developed acute LIMA→LAD anastomotic failure under cardiogenic shock within 24 hours of surgery. Considering the risk of of perforation or avulsion with standard PCI, the team deployed a PK Papyrus covered stent directly across the anastomosis—restoring TIMI‑III flow and myocardial blush successfully.
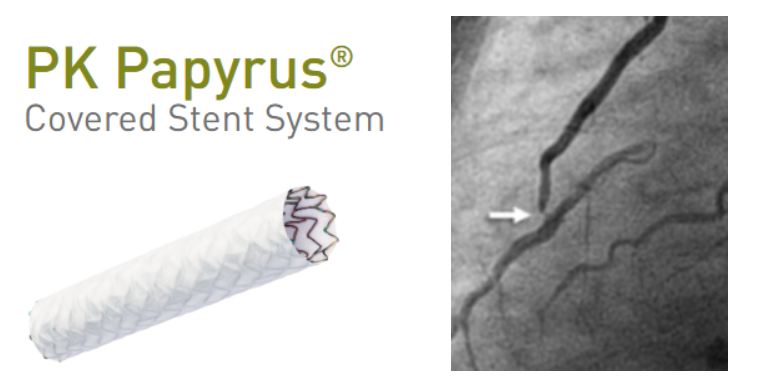
This highlights the covered‑stent’s potential as a first‑line semi‑emergency intervention, offering controlled sealing and avoiding repeat surgery in hemodynamically unstable patients.
What about chronic anastomotic site Lesions ?
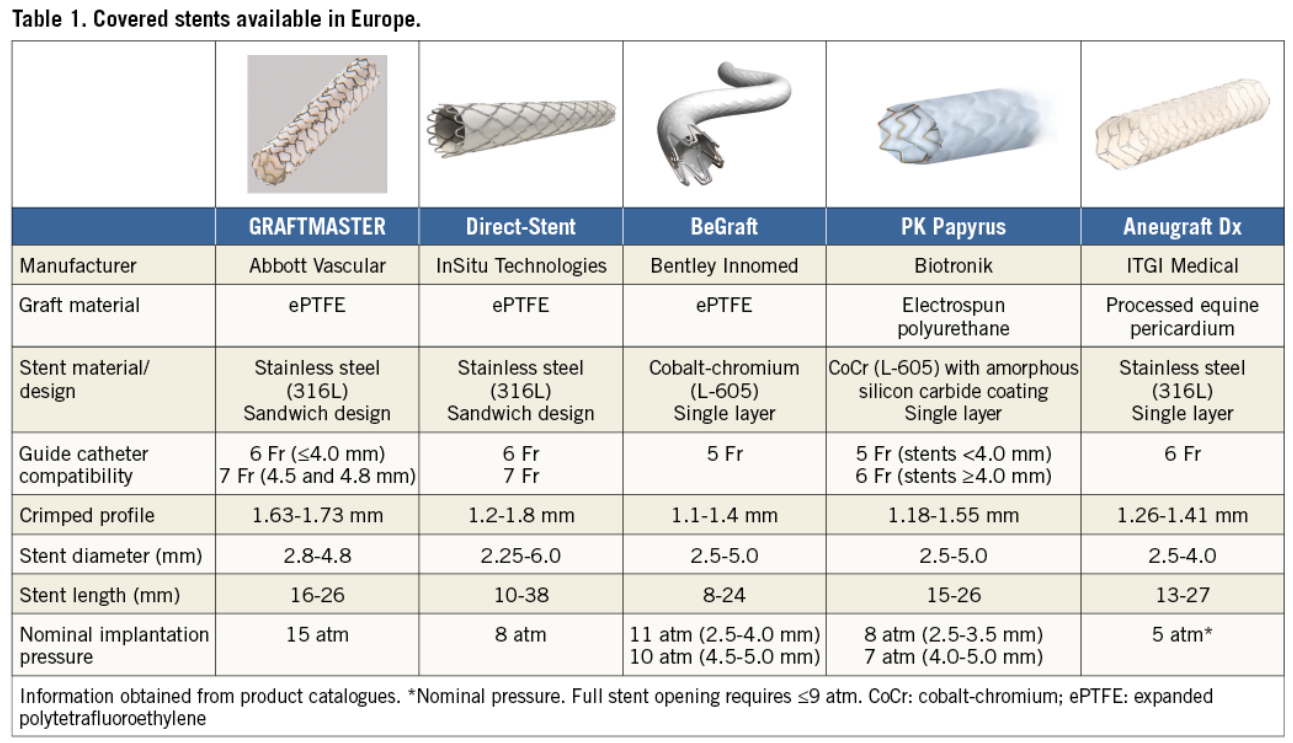
Beyond acute rescue, the covered‑stent may play a valuable role in chronic anastomotic stenoses at the LIMA–LAD junction—lesions notorious for tortuosity and perforation risk. The PK Papyrus platform, with improved deliverability compared to older models like Graftmaster, offers a safer option for such high‑risk anatomical sites . Surprisingly, this indication is absent from covered‑stent guidelines, despite its clear utility in both acute and chronic settings.
Implications of blocking Native LAD Flow
There can be downsides in blocking the native LAD flow by the covered stent. ,However, in reality the proximal flow and to potential branches till the covered stent block is found to be flowing well. In contrary, a key advantage of covering the LIMA–LAD anastomosis is the elimination of competitive flow. In many bypass scenarios, flows between the graft and native vessel compete, potentially compromising graft patency. With the covered‑stent sealing the anastomosis, distal LAD circulation becomes exclusively graft‑dependent, which may actually
- Stabilize hemodynamics by directing full perfusion through the graft.
- Reduce competitive flow dynamics, promoting long‑term graft patency.
- Lower ischemic risk if native LAD disease progresses proximally.
- Finally, the cover acts as a distal protection device against thromboembolic material from proximal friable lesions.
Role of DEB /DES in LIMA to LAD anastomotic lesion
This can be an alternate option. If native LAD flow is considered important and lesion is less complex and risk of perforation is low.

Final message
A simple DOBA (Drug eluting POBA) , or a covered‑stent at LIMA–LAD anastomoses can be a game‑changer, saving lives in emergencies, possibly improving chronic graft outcomes as well. It’s time for interventional cardiology experts to recognize and acknowledge this application, supported by further registry data or trial.
Reference
Posted in acute coroanry syndrome | Tagged anastomotic lesions after lima cabg, biotroniks, covered stent for lima to lad lesion, des in lima, lima interventions, lima to lad pci, newer indication for covered stent, papyrus covered stent, pk papyrus stent, poba vs doba for lima to lad lesion, post cabg lima graft lesions |
Covered stents are exclusively reserved for coronary artery perforations. Yes, that’s what we think. There has been limited exploration regarding the value of covering the complex lesions, which could prevent future coronary events .
It is possible, covered stents might play a extended role , other than perforations as in complex .friable thin capped lesions . As of June 2025 , haven’t found any such study in cardiology literature.
The recently released PREVENT study argued for PCI for patients with vulnerable high risk plaque. Ironically , it is found plaques with very thin cap ie <50microns are at risk of rupture by the radial stress of struts in the immediate or late follow up.
The thought of this study came when we witnessed high recurrent events, due to plaque prolapse, TCFA injury, new plaque ruptures, micro emboli. no reflow etc in patients with complex lesions.
Any past studies done on this aspect ?
There have been some attempts to use covered stents in degenerated venous grafts. Also, the M-Guard stent system was used in the past to seal thrombus during primary PCI. Both showed mixed results. (Gracida 2015)
Are we ready for a trial with a far fetched Imagination ?
What about jacketing and sandwiching the coronary lumen internally with a synthetic layer of tissue? That can potentially prevent recurring events indefinitely. (It is like making a native coronary artery into a Teflon-coated tube.) The proposal may look crazy until we find a inert layer of synthetic tissue to false roof the coronary lumen. But someone can make a start.
Final message
Covered stents are not just meant to arrest blood leaking outwards, in case of perforation , it can also be used seal high risk plaques, that ruptures and leaks its content into the lumen.
*In the following document, a brief outline and proposal is written about such a study. Whoever wants to do such a study, may use it. I wish I could be an external adviser, as I am no longer attached to a teaching hospital or research center.
Postamble
Before , we begin such a study, one may look at the long term outcome of patients who had already received covered stents for perforations. This is important because, PTFE’s pro-thrombotic potential and need for additional vigilance is yet to be defined.
Posted in Uncategorized | Tagged cardiology research proposal, cardiology research topic, copy right free cardiology research, drsvenkatesan, fellows research topic dm dnb cardiology, free cardiology research topics, madras medical college, New role for covered stent, PLAQUE PROLPASE, SEAL TCFA TRIAL, TCFA |
Phrenic nerve arises from C3, C4, C5 cervical spinal nerves ,but essentially from C4 . In the neck, it runs along the anterior scalene muscle, deep to the pre-vertebral fascia. It Enters the thoracic inlet posterior to the subclavian vein and anterior to the subclavian artery.
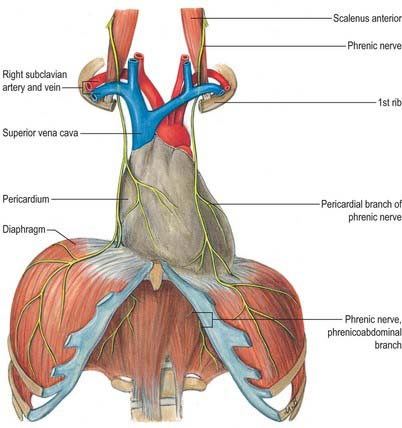
Does It traverse the Pericardial Space?
Contrary to my longstanding belief, realized just now, the phrenic nerve does not enter the pericardial cavity. Rather, It courses within the fibrous pericardium, between the fibrous pericardium (outer layer) and mediastinal pleura. Hence, it is extrapericardial but intimately related to the fibrous pericardium. (Yes, I was indeed a prof of cardiology, teaching students. Wish, I could learn cardiac anatomy from the scratch again)
Anyway, the fact that it runs outside the pericardium, doesn’t give any comfort to the electrophysiologists, both during epicardial and sub-endicardial ablations. It is worth noting the important differences in the course of right and left phrenic nerves.

Difference between right and left phrenic nerves anatomy

Understanding the anatomy of the phrenic nerve is crucial for both cardiac surgeons (of course they see with their eyes) and electrophysiologists. Phreni nerve injury or ablatio can lead to serious consequences.
Right phrenic nerve
Familiarity with phrenic nerve anatomy is key during an ablation. Specifically, the right phrenic nerve should be carefully delineated during endocardial ablation at key sites, such as SVC, the postero-lateral aspects RA. right superior pulmonary vein, and the junction of the IVC and RA. Fortunately right phrenic nerve never cross over the free wall of RV, unlike the LV,
Left phreic nerve
The left phrenic nerve, on the otherhand, should be localized when performing endocardial ablation near the LAA, ablation of left sided accessory pathways,and epicardial ablation of left ventricular tachycardias
How to avoid phrenic nerve injury during RF ablation ?
There are a variety of ways to displace the phrenic nerve from the ablation site, like fluid, air, or balloon inflation. Here is a step-by-step review article in the Journal of Cardiac Electrophysiology in the current issue, June 2025. It is free access too.
Reference
Posted in Anatomy of heart, cardiac anatomy | Tagged cardiac antomy, cardiology anatomy, phrenic nerve anatomy, relationship between phrenic nerve and pericardial space, right vs left phrenic nerve anatomy |
Paroxysmal nocturnal dyspnea and orthopnea are cardinal symptoms of heart failure. The difference between the two has been extensively discussed and debated in medical literature. The key difference is in the time lag that occurs in PND , while orthopnea occur immediately. However, we never looked into PND & Orthopnea with reference LV, RV or biventricular failure.
The fact that Orthopnea occur immediately, raises many critical queries.
It is presumed that the increase in venous return in a recumbent posture immediately causes lung congestion and stimulates pulmonary receptors (J or non-J?) which results in dyspnea. The fact that orthopnea is relieved by sitting posture demands still more explanation. Is it volume-dependent lung congestion, or volume and stretch-dependent RV mechanic receptor stimulation? (or both) I think it is difficult to answer that question.
We get some indirect clues in bed side, by experience. In many patients with Chronic RV dysfunction , orthopnea seems to be less, making it likely pulmonary origin. At the same time, if RV dysfunction is new or acute, it is the raised RVEDP, that is responsible.
Now , we have a problem . Is orthopnea related (more )to RV or LV dysfunction ?
It can have complex inter dependent relationship. In fact, the degree of pulmonary hypertension, the septal push (Reverse Bernheimer effect ) can further confound. Severe RV dysfunction alters the V:Q ratio of lungs, and a also a mismatch between RV vs LV stroke volume.
Final message
The origin of Orthopnea is determined by the status of both RV and LV function. They can either congest or decongest the lung. Realize, in a severely dysfunctional biventricular failure, it is the fine balance between them that keeps the lung dry or wet.
The importance of RV mechanoreceptors and their pathways to dyspnea centers are less understood. While the mechanism of orthopnea is intertwined between the functions of the two ventricles, PND is fairly specific for acute elevation of LVEDP and resultant alveolar interstitial edema. Mind you , orthopnea can occur with totally dry lungs, if its origin is from RV, while it is a rarity in patients with PND.
Post-amble
Time lagged Orthopnea : A proposal for new clinical entity.
We have also seen patients with RV dysfunction mimic PND when they develop dyspnea say 15 to 30 minutes after lying down. Fellows should go back in time and try to re-look and analyze gaps in our understanding of cardinal symptoms.
A small study is easily possible about the incidence of PND and orthopnea in patients with cardiac failure with reference to right and left ventricular function.
Posted in Uncategorized | Tagged clinical cardiology, dyspnea mechanism, mechansims of pnd, paroxysmal nocturnal dyspnea, pnd vs orthopnea, relationship between RV function vs orthopnea |



How can we use AI as a tool of knowledge distillation ?
Here is a deep discussion with Grok 3, on the merits, limitations & validity of DANAMI 2 and PRAGUE 2 , the two old studies on pPCI. Curiously , we don’t have any other studies to quote. As on 2025 , superiority of pPCI hangs precariously on these two decade old studies, which has some serious omissions in the primary end point and its Interpretation. To get into the facts , please go through the following link.
https://grok.com/Is primary PCI really superior to lysis in a global perspective /
It is a long chat, I am sure most of you can’t spare your vital time. But, the truth comes out only at the fag end of the conversation.
Posted in acute coroanry syndrome, acute coronary syndrome, Medcal research, Medical education, Medical ethics, Primary -PCI, Primary PCI, Thrombolysis | Tagged acc aha esc scai criteria, captim trial, cardiology, danam 2, defining success in pci, drsvenkatesan, ethics in medicine, golden hour, medical ethics, nejm, nstemi, pharmaco Invasive strategy, prague 2, pre hospital lysis, ptca, stemi, stream trial, successful primary PCI, symptom to balloon vs symptom to needle time, symptom to door vs door to needle time, time windows in STEMI |

Final message
Since, we can’t sue science for its impurity , or a misbehaving bacteria , an inadequate imaging machine or manipulated health care system,poor doctors have become, the only visible targets.
Related topic
Martin A Makary BMJ 2016;353:i2139 doi: 10.1136/bmj.i2139
Next query in medical ethics
What is the permissible level of error rate, for a medical professional?
Governments can err, Courts can err, pilots can err, meteorologists can err, sportsmen can err, but …
Wish , the prestigious British journal of medical ethics write a position paper on this.
Posted in Uncategorized |
Categories
-

-
The contents of the this blog is being published as Kindle E book , as per the request of many of the readers. Every article will continue to be open source in this site. Again I shall reiterate the book format is not aimed at any commercial intent. It is only to facilitate learning in a single book format Here is the link to book
https://amzn.in/d/euhL5vu Archives
- March 2026 (5)
- February 2026 (8)
- January 2026 (8)
- December 2025 (11)
- November 2025 (7)
- October 2025 (8)
- September 2025 (7)
- August 2025 (9)
- July 2025 (10)
- June 2025 (8)
- May 2025 (9)
- April 2025 (7)
- March 2025 (10)
- February 2025 (4)
- January 2025 (9)
- December 2024 (11)
- November 2024 (8)
- October 2024 (10)
- September 2024 (5)
- August 2024 (5)
- July 2024 (6)
- June 2024 (5)
- May 2024 (4)
- April 2024 (7)
- March 2024 (4)
- February 2024 (8)
- January 2024 (6)
- December 2023 (8)
- November 2023 (13)
- October 2023 (14)
- September 2023 (5)
- August 2023 (6)
- July 2023 (10)
- June 2023 (5)
- May 2023 (5)
- April 2023 (4)
- March 2023 (5)
- February 2023 (2)
- January 2023 (7)
- December 2022 (3)
- November 2022 (5)
- October 2022 (5)
- September 2022 (4)
- August 2022 (3)
- July 2022 (9)
- June 2022 (2)
- May 2022 (1)
- April 2022 (2)
- March 2022 (1)
- February 2022 (3)
- January 2022 (7)
- December 2021 (3)
- November 2021 (5)
- October 2021 (8)
- September 2021 (4)
- August 2021 (6)
- July 2021 (6)
- June 2021 (7)
- May 2021 (5)
- April 2021 (4)
- March 2021 (3)
- February 2021 (6)
- January 2021 (8)
- December 2020 (4)
- November 2020 (5)
- October 2020 (7)
- September 2020 (7)
- August 2020 (10)
- July 2020 (6)
- June 2020 (9)
- May 2020 (9)
- April 2020 (5)
- March 2020 (7)
- February 2020 (3)
- January 2020 (4)
- December 2019 (4)
- November 2019 (6)
- October 2019 (3)
- September 2019 (6)
- August 2019 (3)
- July 2019 (1)
- June 2019 (3)
- May 2019 (2)
- April 2019 (2)
- March 2019 (2)
- February 2019 (4)
- January 2019 (2)
- December 2018 (2)
- November 2018 (2)
- October 2018 (2)
- September 2018 (1)
- August 2018 (2)
- July 2018 (3)
- June 2018 (1)
- May 2018 (3)
- April 2018 (1)
- March 2018 (3)
- February 2018 (3)
- January 2018 (1)
- December 2017 (3)
- November 2017 (3)
- October 2017 (3)
- September 2017 (2)
- August 2017 (2)
- July 2017 (2)
- June 2017 (2)
- May 2017 (4)
- April 2017 (3)
- March 2017 (3)
- February 2017 (5)
- January 2017 (3)
- December 2016 (2)
- November 2016 (5)
- October 2016 (4)
- September 2016 (3)
- August 2016 (5)
- July 2016 (3)
- June 2016 (4)
- May 2016 (3)
- April 2016 (6)
- March 2016 (4)
- February 2016 (3)
- January 2016 (5)
- December 2015 (6)
- November 2015 (5)
- October 2015 (8)
- September 2015 (2)
- August 2015 (5)
- July 2015 (7)
- June 2015 (4)
- May 2015 (6)
- April 2015 (5)
- March 2015 (7)
- February 2015 (15)
- January 2015 (8)
- December 2014 (5)
- November 2014 (9)
- October 2014 (7)
- September 2014 (9)
- August 2014 (5)
- July 2014 (11)
- June 2014 (5)
- May 2014 (4)
- April 2014 (5)
- March 2014 (8)
- February 2014 (8)
- January 2014 (5)
- December 2013 (7)
- November 2013 (7)
- October 2013 (14)
- September 2013 (12)
- August 2013 (15)
- July 2013 (15)
- June 2013 (15)
- May 2013 (15)
- April 2013 (15)
- March 2013 (15)
- February 2013 (15)
- January 2013 (15)
- December 2012 (15)
- November 2012 (15)
- October 2012 (15)
- September 2012 (15)
- August 2012 (15)
- July 2012 (15)
- June 2012 (15)
- May 2012 (15)
- April 2012 (15)
- March 2012 (15)
- February 2012 (15)
- January 2012 (15)
- December 2011 (15)
- November 2011 (17)
- October 2011 (17)
- September 2011 (17)
- August 2011 (21)
- July 2011 (20)
- June 2011 (17)
- May 2011 (15)
- April 2011 (17)
- March 2011 (25)
- February 2011 (20)
- January 2011 (20)
- December 2010 (18)
- November 2010 (21)
- October 2010 (21)
- September 2010 (25)
- August 2010 (20)
- July 2010 (10)
- June 2010 (11)
- May 2010 (19)
- April 2010 (16)
- March 2010 (14)
- February 2010 (22)
- January 2010 (18)
- December 2009 (20)
- November 2009 (20)
- October 2009 (3)
- September 2009 (21)
- August 2009 (19)
- July 2009 (12)
- June 2009 (12)
- May 2009 (11)
- April 2009 (15)
- March 2009 (21)
- February 2009 (4)
- January 2009 (12)
- December 2008 (13)
- November 2008 (9)
- October 2008 (22)
- September 2008 (20)
- August 2008 (16)
- July 2008 (14)
- June 2008 (7)
Blog Stats
- 6,633,282 hits
Please give your feed back .
Click below to see who is watching this website live !

- This site will never aim for profit. Still ,this donation link is added at the request of few visitors who wanted to contribute and of-course that will help make it sustainable .

Please Note

