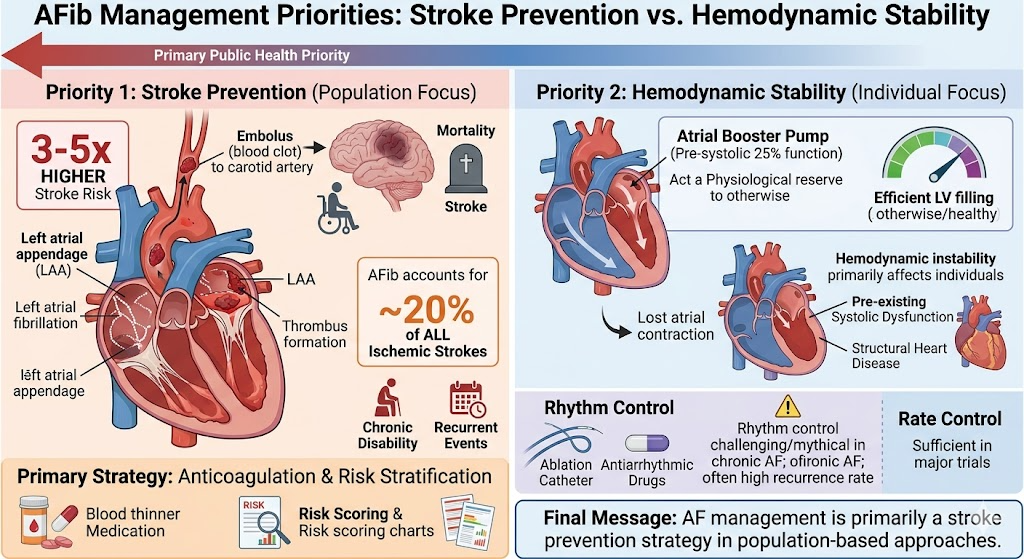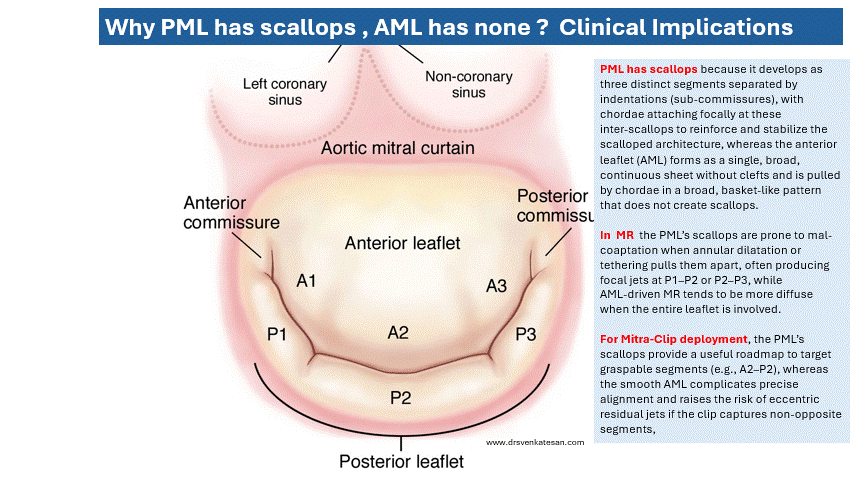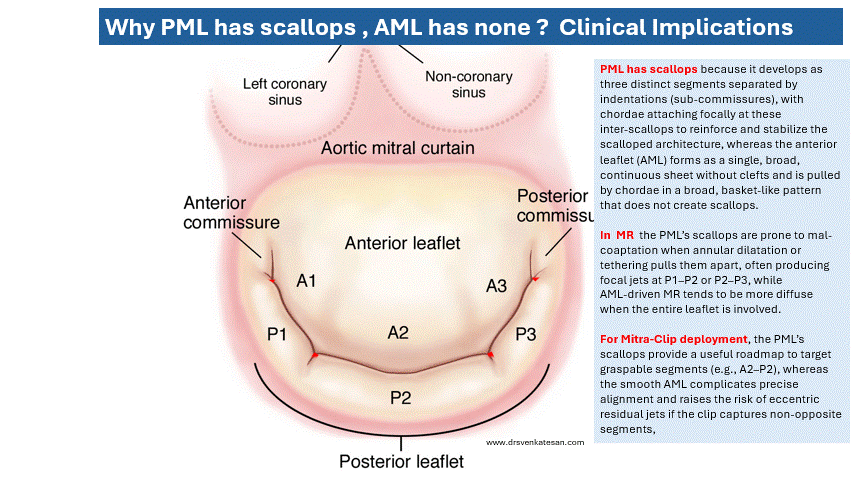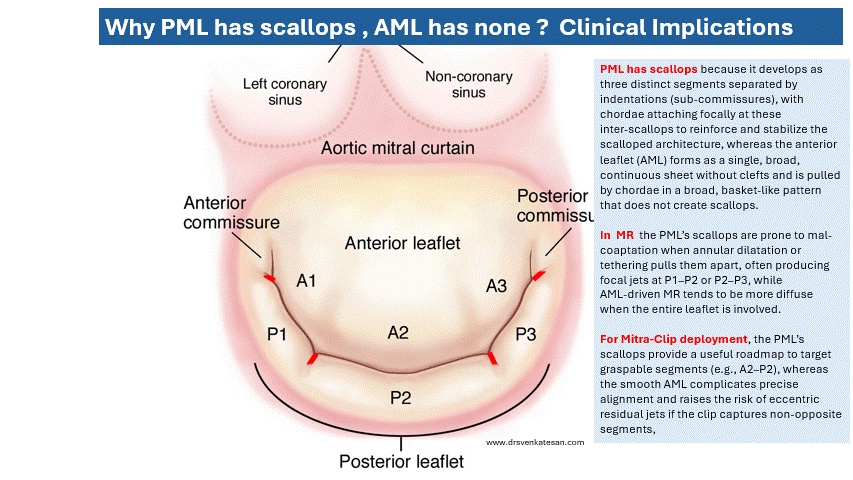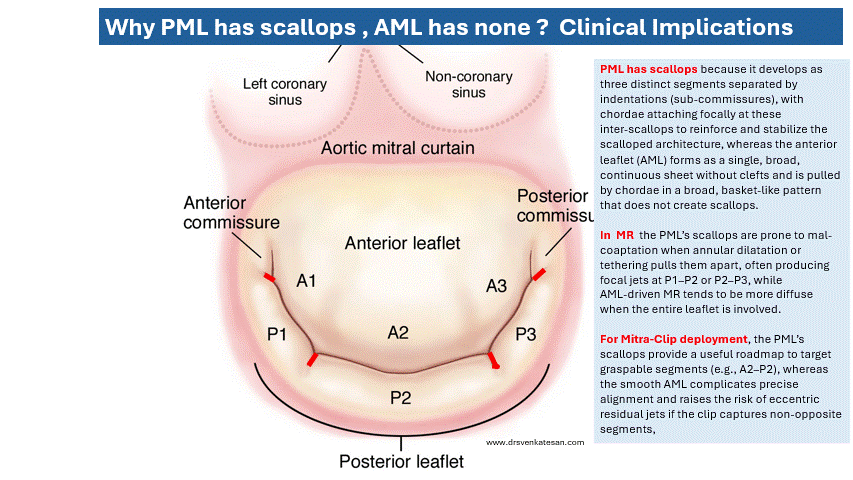

We know south Asians (who constitute about 2 billion people) consistently develop premature and severe atherosclerosis despite largely normal or only mildly elevated LDL-C levels. This “Lipid Paradox” is driven by small dense LDL particles, elevated ApoB, high triglycerides, low HDL, and insulin resistance not classical high LDL cholesterol.
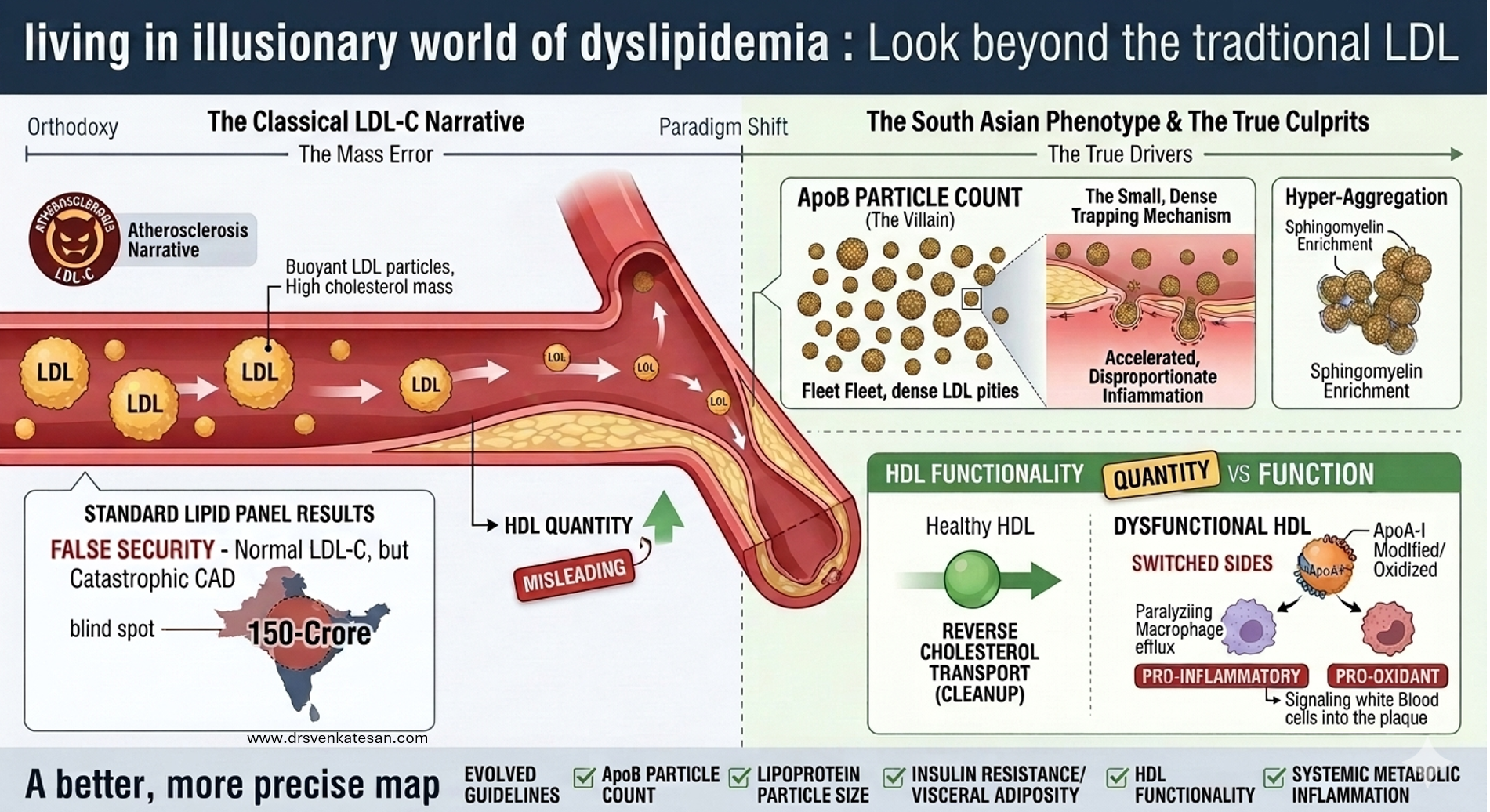
This reveals a fundamental flaw in the current LDL-centric model of atherosclerosis genesis. When one-quarter of humanity develops CAD without high LDL, we must question whether LDL reduction is the primary target or merely a convenient proxy. A broader focus on ApoB, metabolic health, and visceral fat may be more scientifically honest and cost-effective.
Further, HDL molecule has its own problems in being a savior. At a level more than 60 mg/dl, it loses its protective value; rather, excess dysfunctional HDL is harmful as well. Since we have failed to increase HDL by pharmacological means, LDL reduction has gained more attention.(Franczyk B Et all , 2021)
Forget the patient .. target the LDL
Yet, aggressive marketing promotes expensive drugs like, PCSK antagonists, SiRNAs like Inclisiran and ATP citrate blocker Bempedoic acid to target ultra-low LDL levels (<55 or <50 mg/dL) in a population where LDL-C is often a weak tentative target. The latest to join the LDL rat race is the VERVE* 102 yearly Injection , a dramatic temporary RNA editing drug by the pharma giant Eli lilly.
*VERVE-102 consists of a messenger RNA encoding an adenine base-editor protein and a guide RNA targeting PCSK9, which are encapsulated in a lipid nanoparticle incorporating N-acetylgalactosamine (NEJM 2026)
Final message
LDL is definitely one of the culprit in human Atherosclerosis , but it is very difficult to prove , it is a major, universal, risk factor in isolation.This is not mocking the science. This is true at least in our part of the world. To treat a South Asian patient exclusively on standard LDL lowering protocols and projecting it as villain de chief, is not a scientically sound cardiology practice.
Reference
- Volgman AS, Palaniappan LS, Aggarwal NT, et al. Atherosclerotic Cardiovascular Disease in South Asians in the United States: Epidemiology, Risk Factors, and Treatments: A Scientific Statement From the American Heart Association. Circulation. 2018;138(1):e1-e34.
- Agarwala A, Satish P, Al Rifai M, et al. Identification and Management of Atherosclerotic Cardiovascular Disease Risk in South Asian Populations in the U.S. JACC Adv. 2023;2(2):100258.
- Bilen O, Kamal A, Virani SS. Lipoprotein abnormalities in South Asians and its association with cardiovascular disease: Current and future perspectives. J Clin Lipidol. 2016;10(3):543-552.
Postamble
The word “wrong culprit” in the title is intentional. It actually means “not a primary culprit”