Cardiologists have been trying for the last two decades to prove PCI is superior or at least equal, to CABG in multivessel CAD. We desperately needed studies to prevail over FREEDOM and SYNTAX which favored CABG.
FAME series , though never had an intention to compare PCI vs CABG , now we have used the platform to upend it to take on the CABG in multivessel CAD. ( FAME 3)
FAME 1 (Fractional Flow Reserve Versus Angiography for Multivessel Evaluation)
Purpose
The FAME 1 study aimed to compare the efficacy of FFR-guided PCI versus angiography-guided PCI in patients with multivessel coronary artery disease (CAD). The goal was to determine whether using FFR to identify functionally significant stenoses (FFR ≤ 0.80) for stenting, rather than relying solely on angiographic appearance.
Inference
Established that FFR-guided PCI is superior to angiography-guided PCI in multivessel CAD, reducing unnecessary revascularizations and improving outcomes.
FAME 2
Purpose
FAME 2 sought to evaluate whether FFR-guided PCI plus optimal medical therapy (OMT) was superior to OMT alone in patients with stable CAD and at least one functionally significant stenosis (FFR ≤ 0.80).
The study concluded that FFR-guided PCI is beneficial in stable CAD when ischemia is present, reducing the need for subsequent urgent revascularizations compared to OMT alone, though it did not significantly reduce rates of death or MI
FAME 3
Purpose FAME 3 aimed to determine whether FFR-guided PCI using contemporary drug-eluting stents was non-inferior to CABG in patients with three-vessel CAD. The study sought to compare these two revascularization strategies in terms of clinical outcomes, testing the hypothesis that FFR-guided PCI could achieve similar results to CABG by targeting only functionally significant lesions.
Conclusion
At 1 year, FFR-guided PCI did not meet the criterion for non-inferiority compared to CABG for the primary endpoint of MACE (death, MI, stroke, or repeat revascularization), with event rates of 10.6% for PCI vs. 6.9% for CABG.
5 years later in 2025
FAME 3 : 5 year follow up data , released in 2025, tries to confirm the non inferiorty of PCI over CABG in a larger sense.
The study concluded that in patients with three-vessel CAD, FFR-guided PCI is not non-inferior to CABG, with CABG remaining superior for reducing MI and repeat revascularization. However, FFR guidance still refined PCI by limiting interventions to functionally significant lesions.
Did the FAME 3 study compare FFR guided CABG vs FFR guided PCI ?
No, none of the FAME studies (FAME 1, FAME 2, or FAME 3) directly compared FFR-guided CABG versus FFR-guided PCI. Each study had a distinct focus involving FFR-guided PCI, but none incorporated FFR guidance into CABG as a primary comparator. Here’s why this comparison could be meaningless.
Final message
Truths express themselves. We can’t force it to happen.
We know, stress tests can give false positive results suggesting ischemia in at least 20% of patients for various reasons . It can occur with systemic (Anemia) and cardiac conditions such as HT, LVH, baseline ECG changes, or myocardial disease.
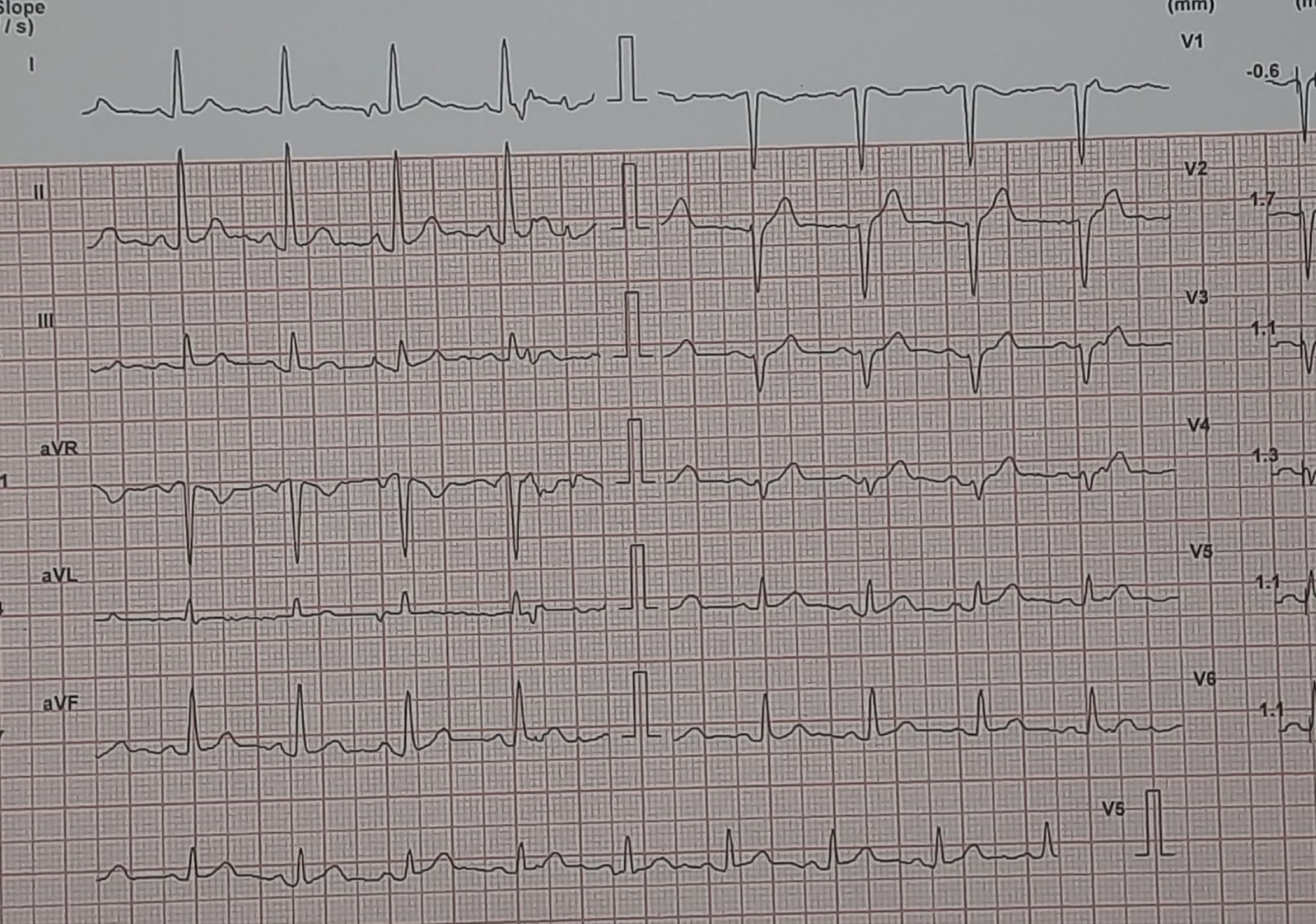
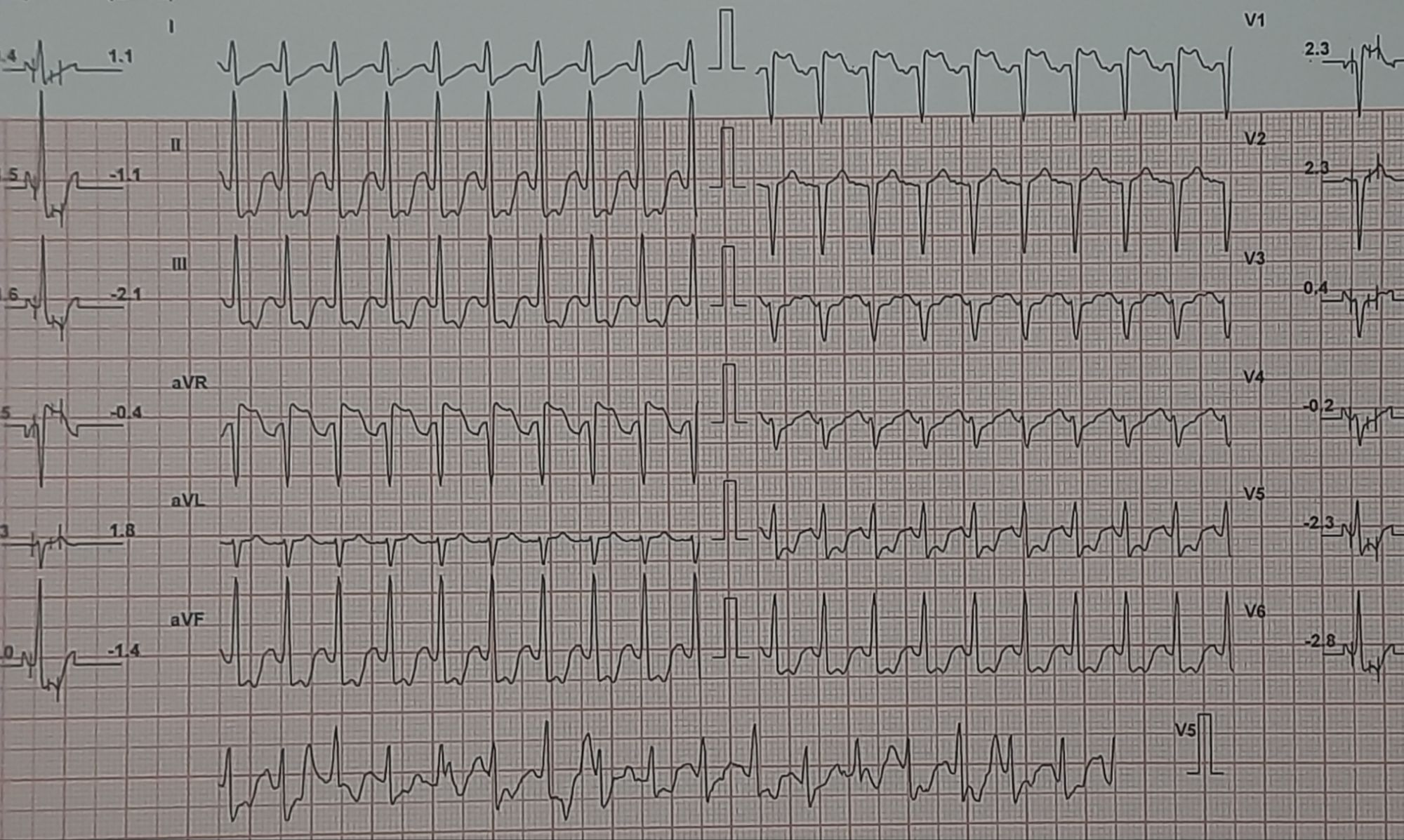
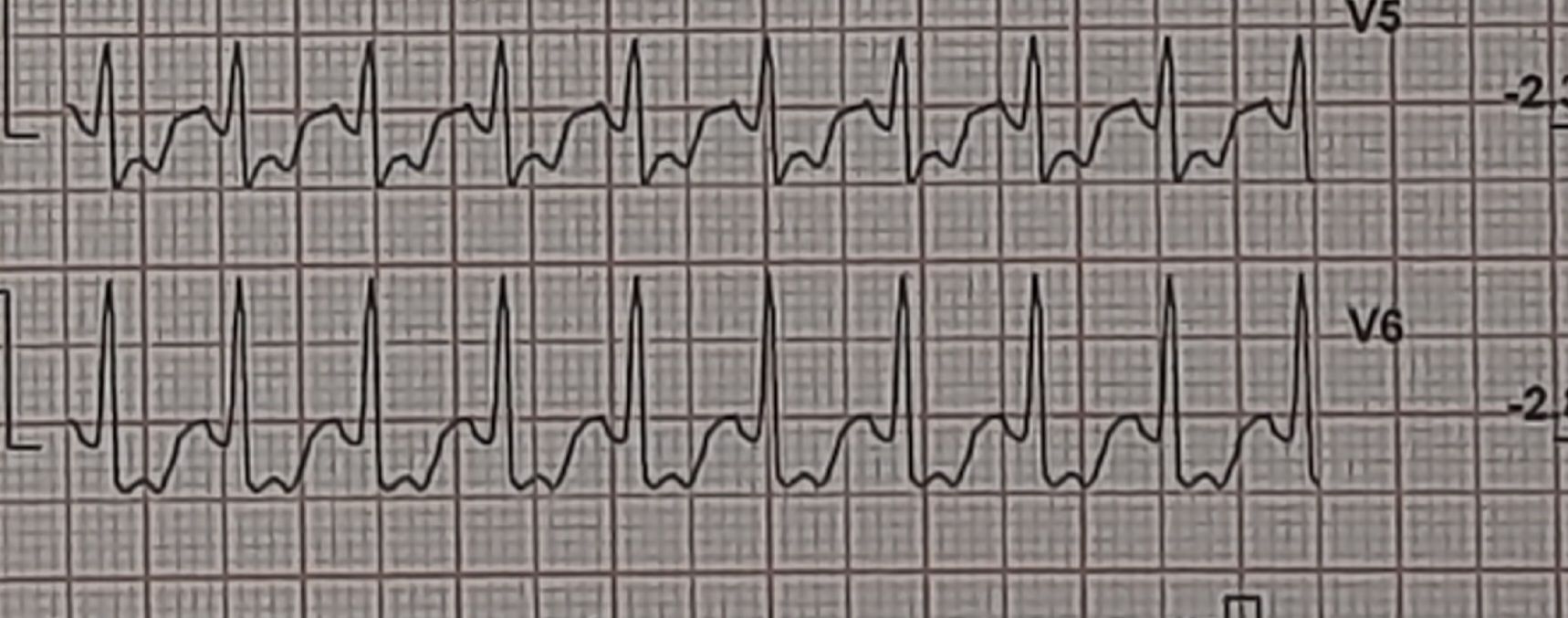
Here is a middle-aged man who went for an annual health check and ended up with this TMT. His exercise capacity was good at 11 METs, stopped at early stage 4 standard Bruce. He was asymptomatic, and every other parameter was normal.
Images: Resting, Peak, severe positive response, in lead V5 and V6. Every cardiologist advised some form of CAG. Opinions were so diverse, ranging between silent left main, tight proximal LAD to innocuous false positive.
What is your inference ? The patient seeked by advice It was indeed an academic stress test. There is a frightening ST depression I said. yes the rest is very likely to be false positive but I don’t have the courage to commit so. Mostly, you can’t escape from a coronary angiogram” . Next option is CT angiogram, Thallium or dobutamine stress.
It was indeed an academic stress test. There is a frightening ST depression . Very difficult to Ignore. May be, it could be false positive but I don’t have the courage to commit so. Mostly, you can’t escape from a coronary angiogram” .Other options are CT angiogram, Thallium or dobutamine stress.
He smiled and said, “You are absolutely right, doctor. Out of 5 cardiologists I consulted, 4 asked me to go for an immediate angiogram. Still, I escaped because of one Egyptian cardiologist.”
I was eager to see what he did . This is the test he did.
Yes. It was indeed a smart move. The shrewd cardiologist did a bicycle ergometry and simultaneous echocardiogram without any drugs or injections. He could confidently rule out significant CAD (by absence of any wall motion defect). Hats off to him. Lets earn some courage from such truely learnt cardiologist.
Final message
Most of us (Cardiologists) find it difficult to trust the physiological data that come from history, ie excercise capcity . We are obsessed with anatomy. Though, we eloborately debate about physiology-based intervention inside cathlab in every conference.
Our flawed intellect keeps asking this question: How can I trust physiology (Flow) without documenting a good anatomy? In fact, truth is the other way around. A good epicardial anatomy rarely guarantee good physiology. (It is worth recalling, CAG, the investigation we celebrate as the gold standard, images only about 2% of the entire coronary vasculature.)
A well-documented near physiologically flowing coronary circulation, negates the need to document anatomy through whihc it flows,however shabby or good it may be. (For the FFR & iFR guys, it must be mentioned that a negative stress test implies a net combined three-vessel FFR of > 0.9.)
Postamble
There was a well-accepted holistic, yet scientific concept roaming around in cardiology academic circles in the 1990s. (Of course, now it is thrown to the dustbin.) It said, if anybody crosses 10 METs in TMT, he or she is unlikely to harbor a significant lesion; even if there is one, it usually doesn’t require a metallic fix.
In pregnant women with significant heart disease : A quick LSCS or a potentially prolonged natural delivery,which is more safe ?
In pregnant women with significant heart disease, the choice between natural vaginal birth and a cesarean section (LSCS) depends on several factors, including the specific type and severity of the heart condition, the overall health of the mother and fetus, and the recommendations of a multidisciplinary medical team (typically involving obstetricians, cardiologists, and anesthesiologists). There’s no one-size-fits-all answer.
Hemodynamics of normal delivery
Natural delivery involves the physiological stress of labor, which includes increased cardiac output, blood pressure fluctuations, and oxygen demand, peaking at 50-80% above baseline during contractions and pushing. For women with significant heart disease (e.g., severe mitral stenosis, pulmonary hypertension, or cardiomyopathy), prolonged labor could strain the heart excessively, potentially leading to decompensation, heart failure, or arrhythmias. The unpredictability of labor duration is a key concern, as it might delay intervention if complications arise.
A planned C-section, on the other hand, offers a controlled environment with predictable timing and monitored anesthesia (often regional, like spinal ,graded spinal or epidural, which can minimize hemodynamic shifts compared to general anesthesia). It avoids the prolonged cardiac stress of labor and allows immediate access to surgical intervention if needed. However, it’s not risk-free: C-sections increase the risk of bleeding, infection, and fluid shifts post-delivery, which can also challenge a compromised heart. Recovery might also be tougher on someone with limited cardiac reserve.
Evidence leans toward individualized decisions rather than a universal preference. For example, women with conditions like Marfan syndrome and aortic dilatation (>4 cm) or Eisenmenger syndrome often have C-sections recommended due to the risk of aortic rupture or acute heart failure during labor. In contrast, women with well-managed, less severe conditions (e.g., repaired congenital defects) might safely attempt vaginal delivery with careful monitoring and a low threshold for intervention.
The American Heart Association and European Society of Cardiology guidelines suggest that for high-risk cardiac patients (e.g., WHO Class III or IV), a planned C-section under controlled conditions is often safer, especially if labor could become prolonged or unpredictable. Epidural anesthesia and assisted vaginal delivery (e.g., forceps) can sometimes bridge the gap, reducing labor strain while avoiding surgery, but this requires a stable patient and skilled team.
Is LSCS really more safe ?
Ultimately, “more safe” hinges on the specific case. A quick C-section in a monitored setting often edges out natural delivery for significant heart disease because it minimizes uncertainty and prolonged stress—but it’s not without trade-offs. The medical team would weigh the patient’s cardiac function (e.g., ejection fraction, symptoms), fetal status, and delivery logistics. If you’re asking about a specific scenario, feel free to give me more details, and I can refine the answer further
Dictums can’t dictate
Is the generally accepted dictum, that ceserain section is meant only for obsterical Indication, still valid in most cardiac patients?
It is true , cesarean section is reserved for obstetric indications—like fetal distress, breech presentation, or failure to progress in labor, has historically been rooted in minimizing surgical risks and prioritizing natural delivery when possible. However, in the context of pregnant women with cardiac disease, this principle isn’t universally upheld as a strict rule. For cardiac patients, the decision often shifts from purely obstetric factors to a broader risk-benefit analysis that includes maternal cardiac safety, sometimes overriding the “obstetric indication only” guideline.
In women with significant heart disease, the physiological demands of labor (e.g., increased cardiac output, blood pressure spikes, and oxygen consumption) can pose a direct threat to maternal survival, even if the pregnancy itself is obstetrically uncomplicated. Conditions like severe pulmonary hypertension, advanced heart failure, or critical aortic stenosis carry high maternal mortality risks during labor—sometimes 30-50% in extreme cases like Eisenmenger syndrome. Here, a planned C-section isn’t just about the baby; it’s about keeping the mother alive. This makes maternal cardiac status a valid indication for C-section, challenging the traditional obstetric-only framework.
However, the dictum still holds some relevance for cardiac patients with milder or well-controlled conditions (e.g., WHO Class I or II, like a repaired atrial septal defect with normal function). In these cases, vaginal delivery is often preferred if obstetric factors don’t demand otherwise, as it avoids surgical risks like bleeding, infection, or anesthesia-related complications that could still tax a less-severe cardiac condition. Assisted vaginal delivery (e.g., with forceps or vacuum) and epidural anesthesia can further reduce labor strain, making natural birth feasible and safe.
Current guidelines, from the American College of Cardiology and European Society of Cardiology, reflect this changing perception . They recommend individualized plans rather than blanket rules. For high-risk cardiac patients (WHO Class III or IV), a C-section is frequently favored—often scheduled around 34-36 weeks if preterm delivery is tolerated—regardless of obstetric status, because the controlled setting trumps the unpredictability of labor. For lower-risk patients, the obstetric indication rule still guide us, unless cardiac monitoring suggests otherwise.
It must me emphasised , the discipline of the multidisciplinary team , especially the egoless ineractions of all members is the key. Type of anesthesia and their cooperation and expertise will be a defining factor many times.
Final message
So, the dictum is no longer valid in all cardiac patients” anymore—it’s just sort of entered our minds and refuse to go away. (There are set of contions and absolute indication for LSCS in heart disese. Every one agrees on that) The purpose of this write up is to look beneath those established Indications.
There is an urgent need for some “academic tinkering” to this decades old, much revered dictum, for the beenfit of mother and baby May be , It applies where cardiac risk is low and obstetric needs dominate, but for severe heart disease, maternal cardiac indication alone can justify a C-section. The shift reflects better understanding of cardio-obstetric interplay and prioritizes outcomes over tradition.
Counterpoint
Guidelines are still dilly-dallying between choices of delivery , based on tradition, technology, expertise & experince (Ref 2 : vouch against LSCS), I think, the obstetrician who is the captain of the multidisciplinary team along with her anesthetist and intensivist are the best persons to take the call. Cardiologist’s role is generally minimal in most situations except for that critical moral support , few management advices and ofcourse for legal protection.
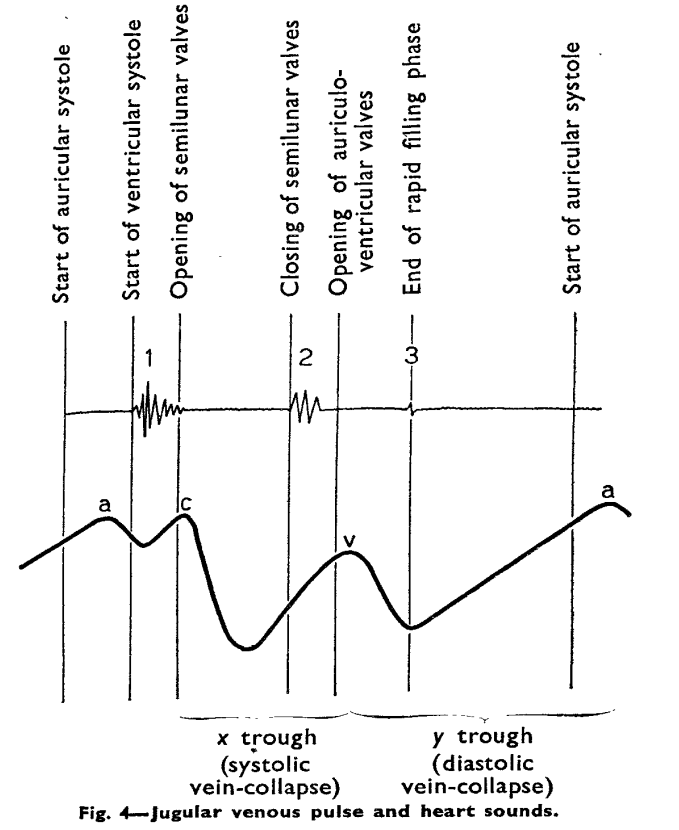
This is the Image of JVP wave forms from the famous original paper by BORST JG, MOLHUYSEN JA. in 1954 paper in Lancet.(Ref 1)
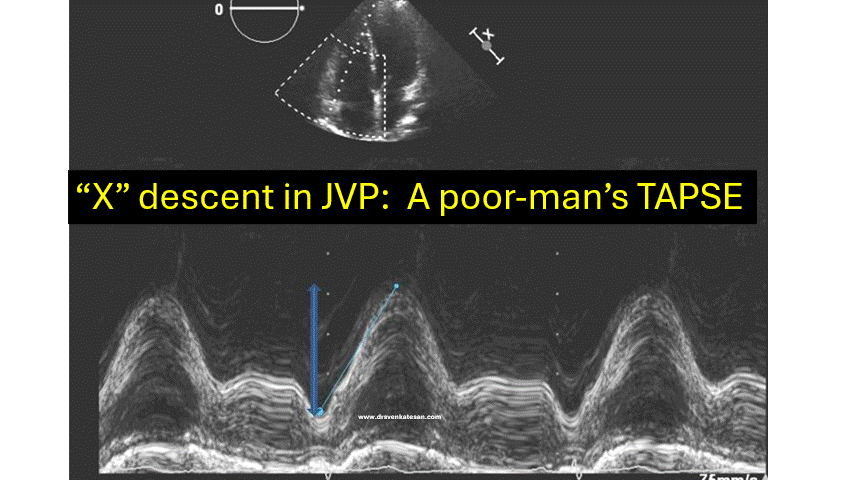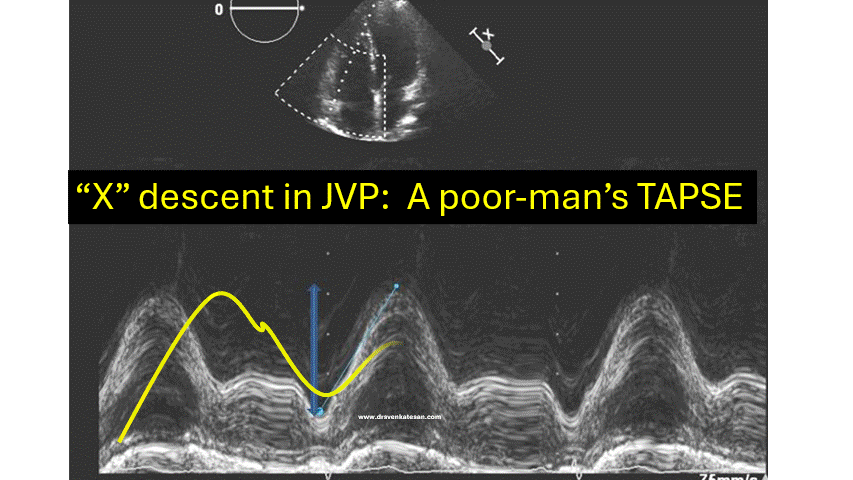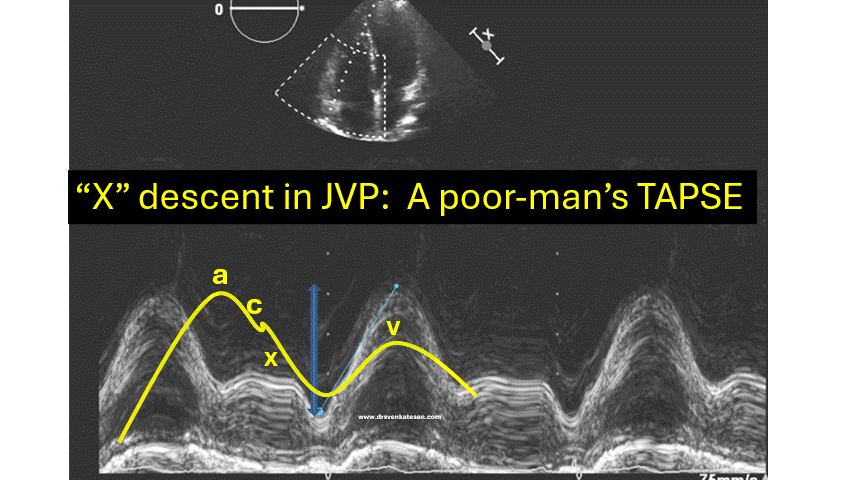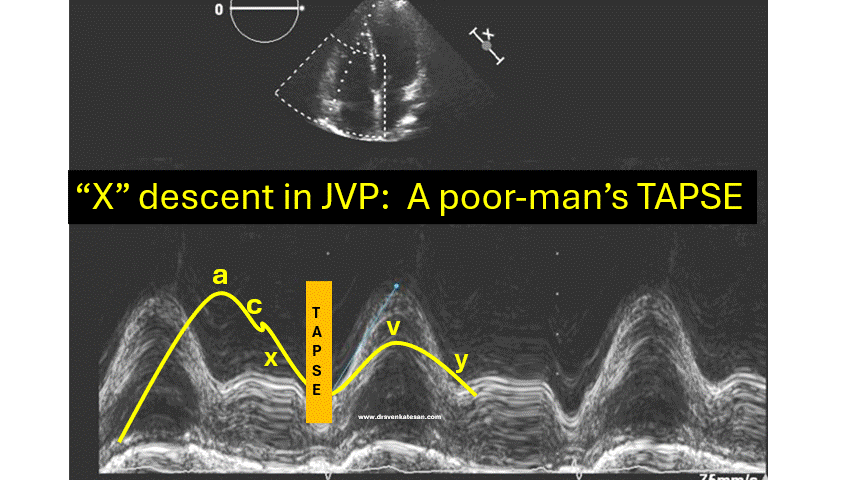
JVP typically has three positive waves and two negative waves. The “A” waves are due to atrial contraction while V waves are due to passive atrial filling. A waves are timed prior to S1 and V waves peak around S2. A tiny c wave interrupts the “x” descent . The word “c” could refer either to the RV contractile force or carotid contamination in the neck or simply a controversial wave.
The downward waves are X and Y descent. The major X descent is due to systolic atrial filling*, when the tricuspid valve is closed. Y descent is diastolic atrioventricular filling.
One interesting echocardiographic correlation has been observed. The force, power, and amplitude of X descent indirectly reflect RV contractility, and it can be referred to as poor man’s TAPSE.
One clinical question often asked in cardiology boards for fellows.
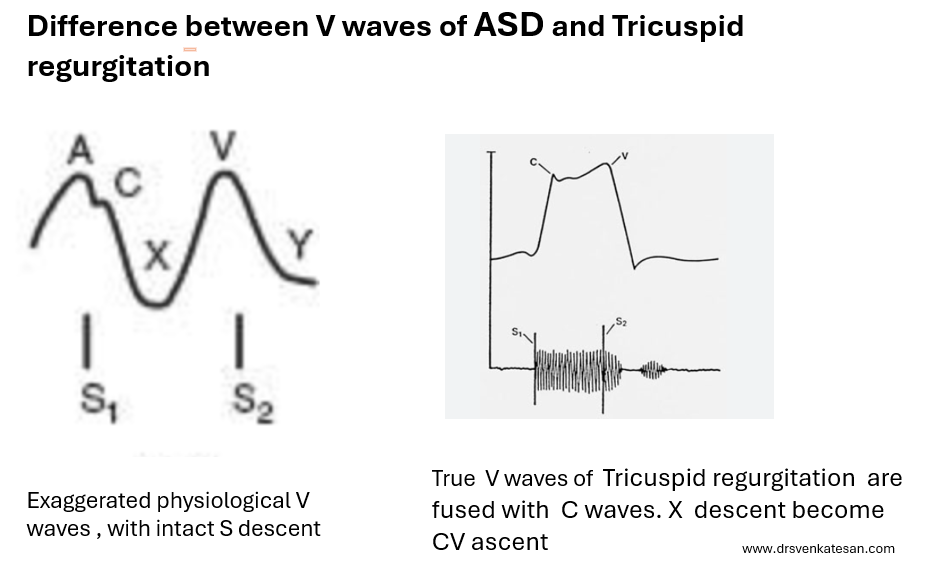
What are the difference between V waves that occur in ASD and Tricuspid regurgitation ?
V waves in ASD vs Tricuspid regurgitation
V wave is due to passive filling wave of atria when the ventricle is contracting and Tricuspid valve is closed.This physiological v wave . In ASD*, this wave just gets exaggerated as the right atrium receives the shunted blood from left atrium when the trisupid valve is closed. Since it almost resembles normal atrial flow pattern , both X descent and Y descent are retained ,and y may be slighly prominent in ASD.
In Tricuspid regurgitation , the V waves are truly pathological in terms of opened tricuspid valve and timing of TR jet which fills the atria in systole rather thanin diastole. (Note this is different from the excessive diastolic filling of atria as in ASD )
While Y descent is prominent in both ASD and TR ,the X descent in TR is lost for simple reason. tricuspid valve is leaking and TR jet abolish the systolic X descend, rather it becomes a X-ascent (Conventionaly called CV waves)
*Please note, the v waves of ostium primum ASD, may not follow this rule as MR from cleft mitral valve further modifies the v wave.
Final message
When we analyse the V waves in JVP , it is important to assess its timing, relative to 2nd sound and also the both the descents to derive maximum hemodynamic information.
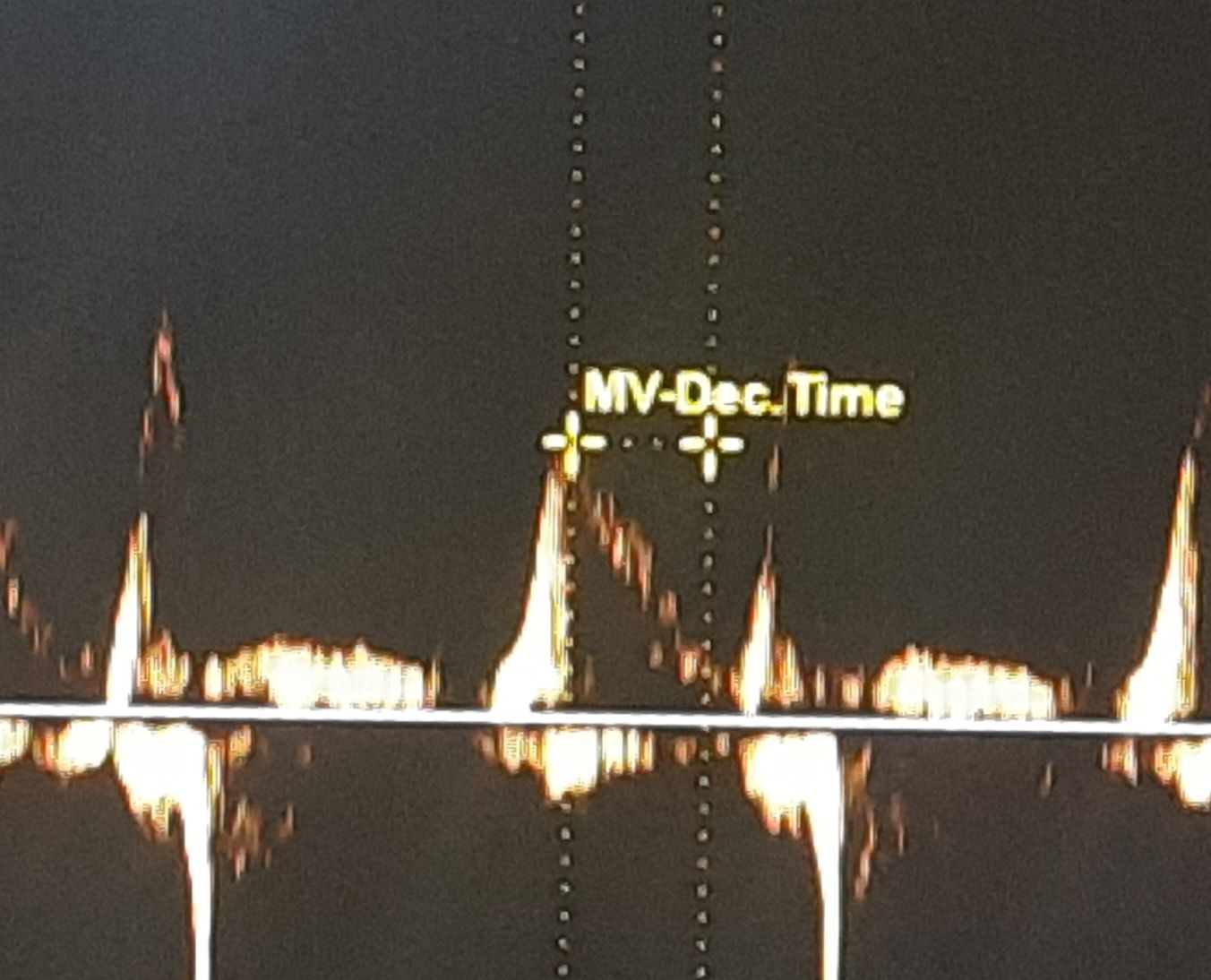
I saw two patients recently, with a similar degree of hypertension and LVH. One with a normal-sized LA and the other with a mild LA enlargement.
When checked for the “E” declaration time, it was found to be absolutely normal in the patient who had LAE. The one with normal LA size had a relatively short DT and his functional capacity was less.
52-year-old man with HT, and LVH with mild LAE. His E DT was very much normal a1 178 ms. He has a good functional capacity. I expected a grade 2 diastolic dysfunction. But, none of the other parameters were convincing. Used to think, if LA is enlarged, it must be a little advanced form of diastolic dysfunction. Though It is still true in many, but, this case, demand us to dwell into these two important parameters of LV diastolic function.
What is the relationship between Left atrial size and Mitral “E” decceleration time ?
The conventional and straightforward answer is they are inversely related.
We know Left atrial size typically reflects the chronicity of elevated left atrial pressure or volume overload, which can result from conditions such as mitral valve disease, left ventricular dysfunction, or atrial fibrillation. An enlarged LA is often a marker of prolonged stress on the atrium due to increased filling pressures or impaired left ventricular relaxation.
Mitral E velocity deceleration time (DT) is a measure derived from Doppler echocardiography, representing the time it takes for the early diastolic filling velocity (E wave) to decline from its peak to baseline.
In healthy individuals with normal LA size and normal diastolic function, DT is typically within a normal range (e.g., 160–240 ms), and LA size does not significantly influence DT. In pathological states, an enlarged LA (e.g., LA volume index >34 mL/m²) combined with a shortened DT (<160 ms) indicate restrictive physiology or advanced diastolic dysfunction.
Question 2
Is this Inverse relation always right ?
There is generally an inverse relationship between LA size and mitral E velocity DT in the context of diastolic dysfunction with elevated LA pressure. LA size increases due to pressure overload, DT tends to decrease. However, the exact relationship is much more complex. If LA enlargement is due to volume overload (e.g., chronic mitral regurgitation) without significantly elevated pressure, DT may not shorten dramatically.
If the LA is stiff and non-compliant, the E deceleration time is likely to be short, and an inverse relation is acceptable logic. But, if the LA is more accommodative and relaxed, mild enlargement actually reduces the LA mean pressure, and E deceleration gets normalized even if it was prolonged earlier due to diastolic dysfunction.
LA behaviour is still a mystery X factor in diastolic dysfunction.
This throws up a fundamental question in our understanding of diastolic dysfunction. Some degree of LA flexibility and compliance reduces the LA mean pressure, and could relieve the symptoms. In this process, the mitral DT also is kept within the normal limits. In fact, now I have asked my fellows to analyze a concept of normalization of DT with progressive LA dilatation in hypertensive patients. This is contrary to the belief that LA dilatation is an ominous sign.
I think it is worth propsoing and pursuing a new concept.” LA dimension has a U curve phenomenon at least within the certain Iniital increments either in size or volume” . LA cannot be too stiff, at the same time it can’t yield out like a balloon.When does an LA decide to dilate and when does it resist is the question ? An agile atria without fibrosis, degeneration, and optimal fluidity extracellular matrix could be the defining factor.
Final message
Understanding the duality in the realtionship between LA size and E deccleration time seems to be crtical. A stiff, non-compliant LA aligns with a short DT and an inverse relationship with LA size in high-pressure states.A relaxed, accommodative LA with mild enlargement may not affect DT significantly and could even normalize it by reducing LA pressure, especially if DT was prolonged due to early LV diastolic dysfunction.
This behavior underscores why LA size and DT must be interpreted along side other factors like LA pressure estimates (e.g., E/e’ ratio), LV compliance, and the underlying pathology.
Detailed answer is also yes : Read further please.
The MitraClip procedure, is designed to reduce mitral regurgitation (MR) by approximating the mitral valve leaflets, can alter the direction or nature of residual MR, including potentially converting a central MR jet into an eccentric one . This possiblity depends on the pre-procedural anatomy, the placement of the clips, and the resulting changes in mitral valve dynamics.
Central MR in ischemic dilated cardiomyopathy (DCM) typically arises from functional MR, where symmetric annular dilation and leaflet tethering (due to LV remodeling) create a central regurgitant jet through a malcoapted valve. The MitraClip works by grasping the anterior and posterior leaflets, usually at the A2-P2 segments, to create a double-orifice valve, reducing the regurgitant orifice area. When successful, this diminishes the overall MR volume, often preserving the jet’s central nature if residual MR remains.
However, if the clip placement is asymmetri or if multiple clips are positioned unevenly, the geometry of the mitral valve can shift. This could redirect the residual regurgitant flow. For example, if the clip is placed more toward the medial or lateral commissure, or if it disproportionately restricts one leaflet’s motion (e.g., excessive tethering of the posterior leaflet), the remaining gap might produce an eccentric jet directed toward the opposite side of the left atrium.
Echocardiographic studies post-MitraClip occasionally report changes in jet direction. While the primary goal is MR reduction, not all procedures eliminate regurgitation entirely, and residual MR jets can appear eccentric depending on how the leaflets coapt after clipping. For instance, if the clip reduces central coaptation but leaves a smaller, off-center orifice, the jet might angle toward the atrial wall, resembling eccentric MR seen in organic valve disease (e.g., prolapse). This isn’t necessarily a conversion from central to eccentric in the classical sense but rather a modification of the residual flow pattern.
Clinical data doesn’t frequently highlight this as a major issue. In trials like COAPT and MITRA-FR, the focus is on MR severity reduction rather than jet direction, and eccentric jets aren’t systematically reported as a post-procedural phenomenon. However, case studies and operator experiences suggest that jet redirection can occur, particularly with suboptimal clip positioning or in complex anatomies.
Implication of new onset eccentric jet
1.Eccentric jet directed towards one of the pulmonary veins can cause unpredictable postural dyspnea.
2.Eccentric jets are difficult to quantify the exact post clip ERV.
3.Can Interfere with favorable remodelling of LA
4.Might Increase IE risk
Final message
Mitra-clip is an innovative catheter-based MR jet interrupter. However, it is not surprising this device could convert a central MR into an eccentric MR, considering the fact that it tampers with mitral valve orifice morphology almost blindly. Adding more complexity is that, the clip brings one more “Neo-regurgitation orifice”. Mitra-clip still can be useful in very selected patients, where it regresses the MR significantly. But, experience tells us the importance of precise clip deployment guided by meticulous imaging and expertise.
Postamble and a follow up question
Can mitraclip convert an eccentric jet into a central one ?
It would be great if this is possible .The problem here is , it need too much precision and overcoming the uncertainity of the iatogenic second jet morphology.
Yes, It’s not a humiliation, to get branded as a new generation cardiologist. In fact, the opposite is true. Sorry, no-blaming any one. We can’t avoid it as well. It is the wages, we are foreced to pay for sensationalised technological sins , that is Imploding in the world of medical science.
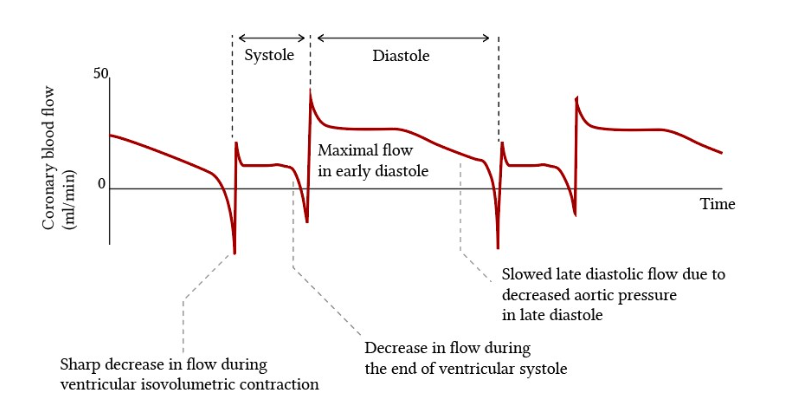
Coronary bloodflow is primarily known, to occcur as a diastolic circulation. Does that in any way mean coronary artery diastolic pressure, can exceed the systolic pressure ?
A. No. diastolic BP can never exceed systolic BP in side the coronary artery.
B. Yes. Coronary diastolic BP is higher than systolic, since there is little blood flow during systole due to myocardial compression.
C. There is not much difference between systolic and diastolic pressures, within the coronary artery . We need to bother only about mean perfusion pressure.
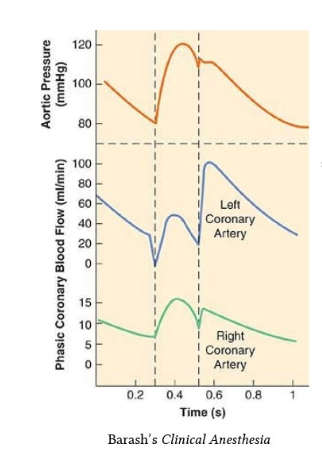
D.It is true, the coronary blood flow is compromised in systole and primarily occur in diastole .Still, the epicardial coronary arterial compresssion is not that significant. Hence systolic pressure blunting is negligible. This is called the pressure -flow paradox.
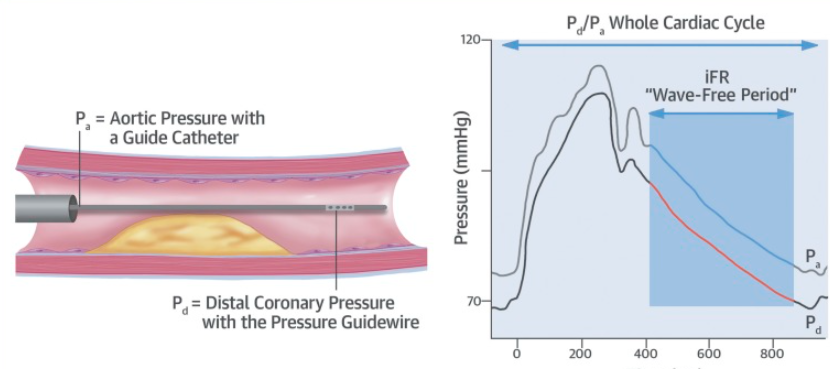
Answer : D (Ref Image 2)
What is the normal intra-coronary arterial pressure in systole and diastole? I could not get a clear answer to this question. Logically it should be sane as in radialartery 120/80mmhg. Surprisingly, most literature discusses only coronary blood flow, which primarily takes place in diastole. (Does that mean the pressure would be less in diastole, so that blood flows easily?) The complexity in understanding intra-coronary pressure , is because, we don’t know the exact blood volume, flow vs pressure relation in this dynamic organ.Further, mechanical force/pressure exerted by the muscle ,can it be recorded , within the lumen , and quntify it sepearately ?
The classical illustrations that are found in cardiac physiology literature about the dominance of coronary blood flow during diastole (Image source Ref 2)
During FFR studies the Intracoronary presssure curves almost mimic radial pulse. No where we could see the effect of mechanical compression . It is likely , the epicardial coronary artery do not get compressed that much , only the micro circulation gets squeezed.
We realise ,coronary perfusion pressure, mean coronary arterial pressure, and coronary arterial wedge pressure are more important than systolic and diastolic pressures . The mean coronary artery pressure is around 45 to 60 mmHg backed up with good autoregulatory mechanism. We are not clear how this autoregulation is modified by lesion tightness. Documentation of true coronary arterial systolic BP in physiology and various pathologies is an important academic vacuum that youngsters can explore.
1.Clinical Implication : Does LV dysfunction has a favorable efffect on coronary perfusion ?
If LV contraction interferes with coronary blood flow, patients with severe LV dysfunction, may gain some advantage as systolic blood flow can happen more easily, and myocardium is perfused better, provided the aortic systolic pressure not too low enough.
2.How common is angina in DCM ? and Why ?
Angina in DCM is an exception despite elevated LVEDP. Is the above logic explain why very few dilated cardiomyopathy patients experience angina? Even in ischemic cardiomyopathy, once it sets in, Intensity of angina is mitigated or completley eliminated.(of course at the cost of failure). Is it nature’s response to prevent angina?
3.Why systemic hypertension is a weak coronary risk factor ?
Unlike the brain, where stroke risk is directly related to systolic BP, fortunately sudden systolic spikes rarely get a chance to attack the coronary endothelium as much of the coronary lumen is relatively closed (? to be confirmed , atleast during rapid ejection phase of systole) In this context, we can also be happy there is no risk of myocardial hemorrhage due to HT. However, there is evidence that diastolic BP carries much risk for CAD, as do Isometric exercises when DBP exceeds out of proportion to systolic BP.
4.Differential intra coronary pressure , RCA VS LCA is well knwon asthe RV contraction is not good enough to compress the RCA.This adds a new hemodynamic concepts in RCA CAD.(We have done a study where we found thrombolysis was more effective in RCA apparently due to bi-modal continuous delivery of the lytic drugs, unlike the left system)
5.During CPR , what would be coronary hemodynamics of chest compression ?
When we compress, it is systemic systole, and when we release it becomes coronary diastole. In fact there is now evidence to suggest , too rapid and hurried contractions reduce the success rate of CPR. The inter compression time is to be atleast 4 or even 5 full seconds, to enable coronary perfusion.The mean pressure during CPR is to be atleast 40mmhg. (Yannopoulos D et al , Resuscitation. 2005)
Final message
It is surprising why we are not recording intra-coronary pressure directly and trying to understand this. We need to go 100 years back for that Wiggers article in search of truth. (Ref 1). This is an area of good research for cardiology fellows. Also, next time,when you do FFR or IFR, ask this question : Why proximal reference pressure is taken at the aortic root instead of just before the lesion ?
OCT, the magical intraluminal coronary vision, has been a great innovation that helps us to decode many uncertainties in the morphology, behavior and vulnerability of coronary plaques. It is used widely in pre- or post-PCI or even asssit during the implantation of stents. The role of OCT/IVUS is sometimes deemed critical in dealing with left main and bifurcation lesions.
Of course, there were some overuse of OCT as well, as many centers did it for some academic fun, even in some innocuous lesions. Meanwhile, there is a striking miss. We probably failed to accrue the benefits of this revolutionary imaging in the graft evaluation. Its real role could be in LIMA grafts including anastomotic site or SVG lesions in the immediate postoperative or at long-term follow-up lesions. As far as I understand, it is very rare for cardiologists to attempt imaging these sophisticated tools in LIMA or SVG.
Published data on OCT IN LIMA
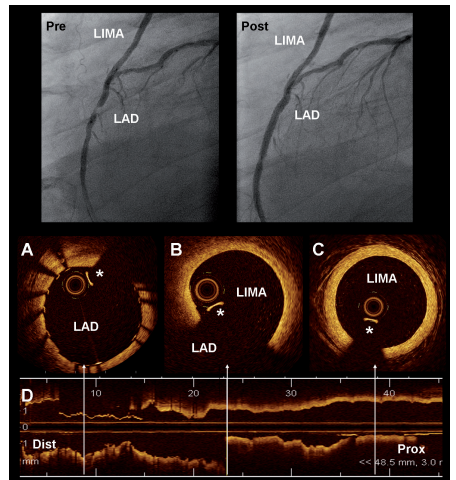
Here is a paper from a stalwart in coronary interventions, Dr. Patrick Serruys and his team from the Netherlands (Published in 2009 ,but surprised to find not many takers)
Image source & Courtesey : Ref 1 Optical coherence tomography (OCT) visualisation of left internal mammary artery (LIMA). The superior panel shows the angiogram of a patient with a graft of LIMA to the LAD.
Why OCT/IVUS is less popular in graft assessment ?
*Graft follow-up often falls under surgeon’s domain. They don’t call for check angio often, unless the patient is really, really symptomatic. (CT angiogram is more popular in post CABG)
*From cardiologist’s point of view, they rarely deem it to be necessary. Reason being, it could be technical (Will the venous graft tolerate the OCT wires?)
*Lack of experience and apprehension
*Lack of publsihed data.
Final messge
It is true, doing regular graft angiogram, by itself is less than 5-10 % of all angiograms . Asking for OCT in that population is big deal .Still, OCT can be a valuable in providing crucial information in the assessment of both LIMA and SVG, at least in the former. One more purpose of OCT is, its offline use to assess the integrity of LIMA graft on table prior to CABG. It can confirm patency and possibly rule out any significant takedown injury, that is missed otherwise.
Though the left atrium is the superior most chamber of the heart , it loses its gravity-assisted LV filling advantage in a lying posture. In patients with compromised heart function, this becomes a symptom defining factor. No surprise, patients during episodes of LVF or paroxysmal nocturnal dyspnea, natural forces make them sit up by default, and bring the LA superior & over the top of LV hence its filling is augmented. One more factor that operates is that, IVC orientation, which assumes slope and reduce venous return velocity. In the process, they decongest the lungs and patient gets Immediate relief. In fact, pillows work faster than diuretics and we can technically call it low-cost LV assit devices.
Note, how the LA takes control by its superior position, when the patient assumes erect posture from supine. In fact ,the number of pillows used, by the pateint has some direct correlation with LA mean and Echo cardiographic E/e ‘ . ESCAPE study suggest a possiblity of correlation of this LVEDP with right sided JVP as well.( Drazner et al Circ Heart Fail. 2008 )
Final message
This post may not be relevant to cardiology fellows. Whenever we receive a dyspneic patient in heart failure, prop them up with few pillows. This lesson is taught right in the first-year clinical rounds. I wanted to highlight the anatomical and hemodynamic basis of the sitting-up posture and its impact on LA mean and LVEDP. By some crazy stretch of imagination, pillows can be referred to as a temporary LV assist device.
Research suggestion for fellows
Some of you can do you a study in cath lab, how much the LA mean pressure is altered with reference to posture. It could appear a flimsy study in this era of TAVR/Mitra clips. Sill, we have an good opprtunity to analyse these things as we enter all chambers of heart in routine fashion for some indication or other. This will make us understand LV filling physiology in a better way. (Recalling the days of Guyton & Rushmer when they strugggled to know computational models to measure the pressure gradients)
A question for our hemodynamic acumen?
How does the LA empty in to LV , when LV inflow conduit need to operate against gravity during head down feet up postion as in many sports like bungee jumping or in some asanas (Shirshasana) . Has any one attempted, to know , how would be the E and A velocity across the mitral valve in this posture .Wish some one take on this and report ,if no one has done it before please add some credit . (Just kidding)
Caution
Patients (even some healthy) with diastolic dysfunction especially in elderly, should never attempt to do such sports or indulge in any compromised posture that brings LA below the LV.
The contents of the this blog is being published as Kindle E book , as per the request of many of the readers. Every article will continue to be open source in this site. Again I shall reiterate the book format is not aimed at any commercial intent. It is only to facilitate learning in a single book format Here is the link to book https://amzn.in/d/euhL5vu
Click below to see who is watching this website live !
This site will never aim for profit. Still ,this donation link is added at the request of few visitors who wanted to contribute and of-course that will help make it sustainable .
Please Note
The author acknowledges all the queries posted by the readers and wishes to answer them .Due to logistic reasons only few could be responded. Inconvenience caused is regretted.