100% occlusion of a coronary artery result in STEMI.This includes both thrombus and mechanical component .We are very much blinded till we touch , feel and see the lesion with a wire or IVUS to quantify the mechanical component’s contribution in the genesis of STEMI.It is generally believed (True as well ) thrombus is the chief culprit .It can even be 100 % thrombotic STEMI with just a residual endothelial erosion and hence
zero mechanical component .However , the point of contention that non flow limiting lesion is more likely to cause a thrombotic STEMI than a flow liming
lesion seems to be biased and misunderstood scientific fact .
What happens once 100 % occlusion take place ?
Sudden occlusion , is expected to evoke a strong fire fighting response within the coronary artery.The immediate reaction is the activation of tissue plasminogen system. In this aftermath few succumb . ( Re-perfusion arrhythmia generated as VF ) .The TPA system activates and tries to lyse the clot.The volume , morphology, attachment, content of thrombus , and the elasticity of fibrin mesh , location of platelet core would determine the life and dissolvablity of thrombus. Even a trickle flow can keep the distal vessel patent .(Please note a timely TIMI 2 flow can be a greater achievement than a delayed TIMI 3 flow !)
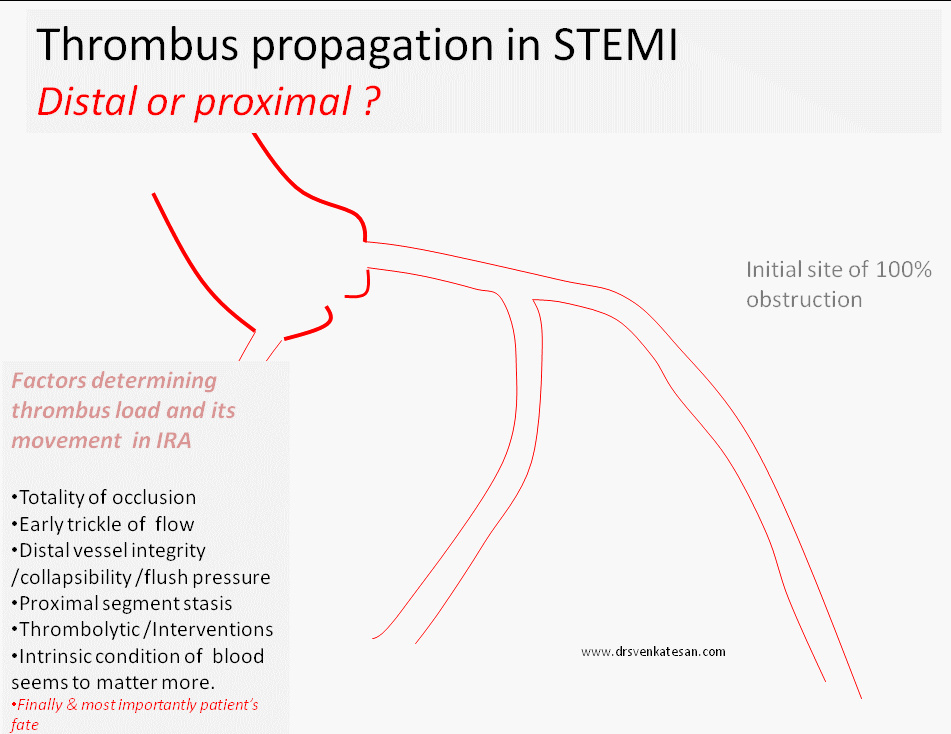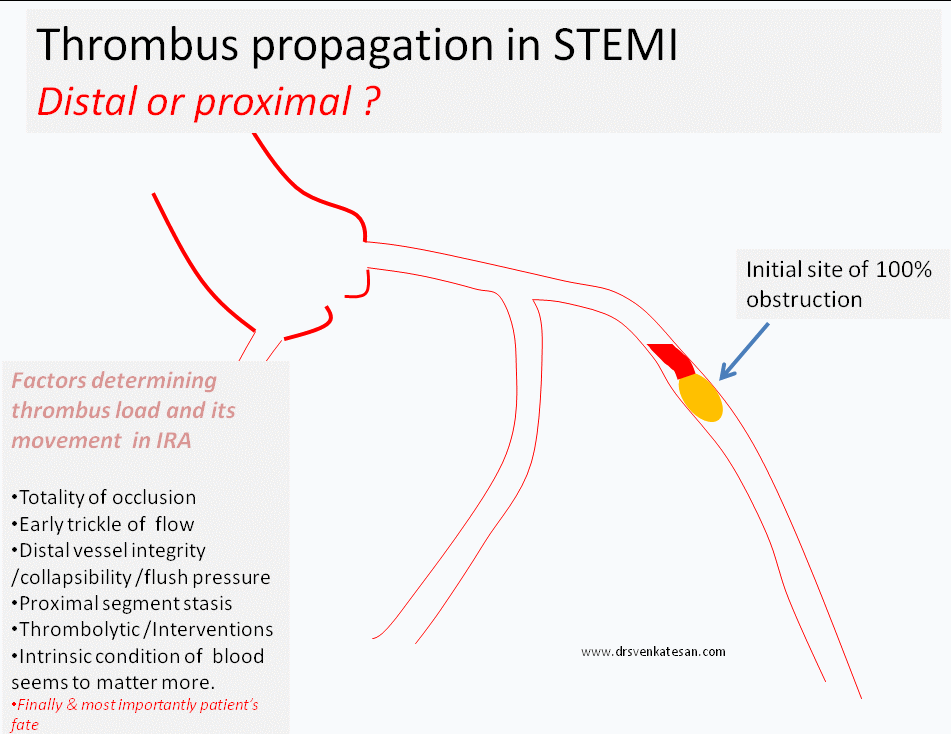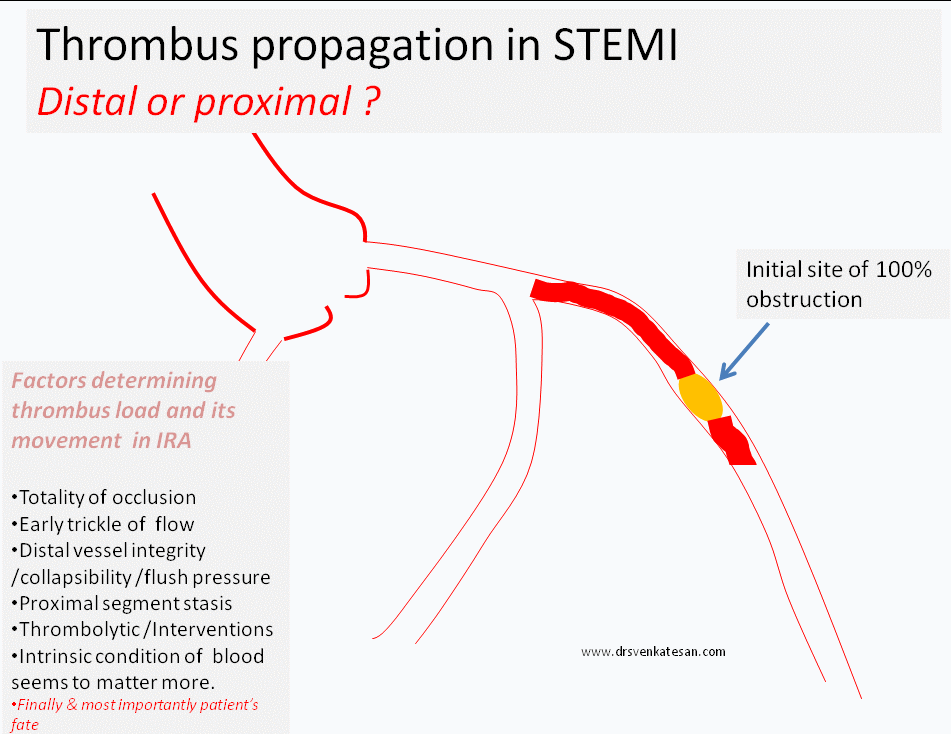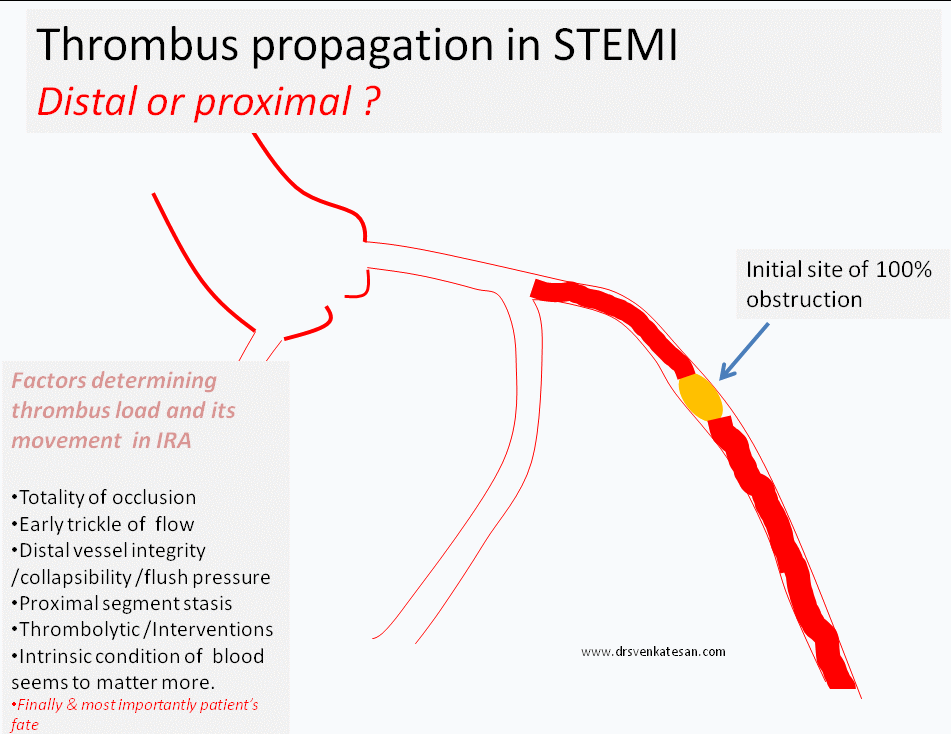
What happens to the natural history of thrombus in STEMI ?
Thrombus formed over the culprit lesion can follow any of the following course
- Can remain static
- Get lysed by natural or pharmacological means
- Progress distally (By fragmentation or by moving en-mass )
- Grow proximal and and involve more serious proximal side branch obstruction
- Organise and become a CTO
Factors determining thrombus migration
The interaction between the hemodynamic forces that push a thrombus distally and hemo-rheological factors that promote fresh proximal thrombus formation are poorly understood. The altered intra-coronary milieu with a fissured plaque covered by platelet vs RBC / fibrin core, totally of obstruction, reperfusing forces , re-exposure of raw areas and the distal vessel integrity all matters.
While, logic would tell us, thrombus more often migrates distally assisted by the direction of blood flow, an opposite concept also seeks attention , ie since the blood flow is sluggish in the proximal (to obstruction site )more thrombus forms in segments proximal to obstruction.
(In fact, its presumed in any acute massive proximal LAD STEMI , it takes hardly few minutes for the thrombus to queue up proximaly and clog the bifurcation and spill over to LCX or even reach left main and result in instant mechanical death.)
What is the significance of length and longitudinal resistance of the thrombotic segment in STEMI ?
If thrombus is the culprit let us get rid of it , this concept looks nice on paper , but still we don’t know why thrombus aspiration in STEMI is not consistently useful. We also know little about the length of the thrombotic segment .When a guide wire is passed over a STEMI ATO it may cross smoothly like “cutting a slice of butter” in some , while in few we struggle and end up with severe no-reflow inspite of great efforts .Why ?
What is the Impact of distal collateral flow in flushing fresh thrombus ?
The efficacy of collateral flow in salvaging myocardium is underestimated. Distal vessel flow if perfused partially by acute collaterals the thrombus load is not only less it’s soft and fail to get organised early that would help cross the lesion easily.




TRANSFER-AMI study : Transfer with caution . . . bumpy roads ahead !
Posted in Cardiology -Interventional -PCI, cardiology -Therapeutics, Cardiology -unresolved questions, cardiology journal club, cardiology journals, Uncategorized, tagged comments about transfer ami, facilitated pci, FAILED THROMOLYSIS, journal watch transer ami, letters to the editor transfer ami, nejm transfer ami, REACT STUDY, rescue pci, routine early pci, stemi, tenecteplase failure, time window for pulmoanry thromolysis, TRANSFER -AMI STUDY on January 14, 2011| Leave a Comment »
Preamble
The much published TRANSFER -AMI study has few important queries to ponder about.It was supposed to test the role of routine PCI following thrombolysis. In other words it compared rescue only strategy with routine strategy.The caveat is , even among failed thrombolysis, the rescue strategy has not convincingly proven superior to medical management (if the time is lapsed ) as much of the damage is done .
Will the investigators share their experience ?
Finally
Why the title of the paper says it is about “Routine angioplasty” and the conclusion emphasizes it is indeed “high risk subsets ofangioplasty” (While the study itself involves a 92 % least risk Killip class 1 ) . Why this double dose of confusion ? (Is it deliberate ! Which i think is unlikely )
NEJM please take note of this . . .
All that glitters are not natural glitter . . .some are made to glitter !
Rate this:
Read Full Post »