
Posted in Uncategorized, tagged artificial intelligence, dr s venkatesan, dr venkatesan sangareddi, future of medical ethics, gene therapy, hippocrates, journal of medical ethics, lown foundation, madras medical college, medical ethics, medical quotes, nano medicine, nejm bmj lancet, noble profession, principles of practice of medicine, quotes medical ethics best, sir william osler on January 28, 2020|
Posted in acute coroanry syndrome, Cardiology -Therapeutic dilemma, Uncategorized, tagged lv dysfunction, primary pci, successful primary PCI on January 23, 2020|
As we try to progress in our knowledge towards absolute truth, we need to admit our errors first. I think, one such error is blinking right in front of us in the vibrant corridors of coronary care and cath labs every day! It is about the definition with which we deal the success of primary PCI. (A supposedly revolutionary acute coronary therapeutics this century)

Waiting for the day . . . when all those fancy primary PCIs that leave the myocardium hurt (& retire ) with significant LV dysfunction to be reclassified as clear cases of primary PCI failures.
Posted in Cardiology - Clinical, Clinical cardiology, Uncategorized, tagged does raised rvedp cause dyspnea, dyspnea in pulmonary hypertension, mechanism of dyspnea on December 18, 2019|
Exertional dyspnea disproportional to the effort is the most common (cardinal)symptom of heart disease. Whenever we discuss the mechanism of cardiac dyspnea , we primarily attribute it to left heart disease, elevated LVEDP and the resultant pulmonary congestion.Conventional teaching in the past (may be in the present too !) doesn’t implicate raised RVEDP in the genesis of dyspnea.
It’s good to recall , the sensation of dyspnea is felt at the peri -Amygdala nuclear zone after complex processing with various cortical and sub-cortical level .It is subjected to as many afferent triggers other than J receptors in pulmonary micro circulation. (Eg Exercising skeletal muscle). It is believed, mechanical stretch receptors exist within the walls of heart along the sub-endocardial aspects of chamber.
(Muscle spindles which are the sensors of muscle tension are extensively noted in skeletal muscle that contribute to the origin of dyspnea .We are not yet accruing enough evidence whether cardiac muscle do have the same muscle spindle or it’s equivalents to cause dyspnea when stretched. However, we clearly witness in the practice of clinical cardiology , isolated elevation of RVEDP ( also RVSP ) to cause significant dyspnea in specific clinical situations.
Potential causes for Isolated Right ventricular dyspnea
*RV diastolic dysfunction is still a Infantile hemo-dynamic concept .Whether it can raise RVEDP significantly during exercise and Independently contribute to dyspnea is at best a hypo-science.
Role of muscle spindle and mechno-receptors
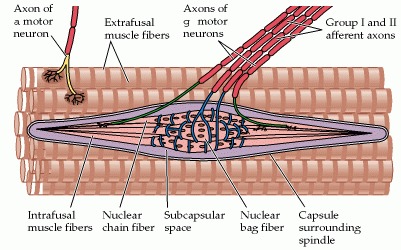
structure of skeletal muscle spindle. Though we don’t have a highly developed spindles in smooth muscle and cardiac muscle we have evidence to suggest cardiac neural ending do have mechano-receptors with afferent connection through visceral neural plexus that can trigger both heart rate and respiratory centers Further reading : Neuroscience. 2nd edition. Show details Purves D, Augustine GJ, Fitzpatrick D, et al., editors. Sunderland (MA): Sinauer Associates; 2001.
Bain-Bridge reflex: The hidden link in right heart dyspnea
Bain-Bridge reflex is a 100 year old concept. still helping us to understand the basics of right heart hemodynamics and how adjustments with acute volume loading take place.He proposed that veno-atrial stretch receptors are located primarily in great veins as it enter ,right atrium (RV as well).
This gets activated through vagus and stimulates in brain-stem sympathetic system and increase the heart rate to handle the excess blood reaching the heart. How often we feel the symptom of palpitation whether due to this reflex ( when it is operating) is not really tested. But, what we can infer is , the surge in sympathetic tone perceived can be perceived as dyspnea.
*Clinical Relevance of the Bezold–Jarisch Reflex and its possible interactions with Bain Bridge reflex is a different topic.
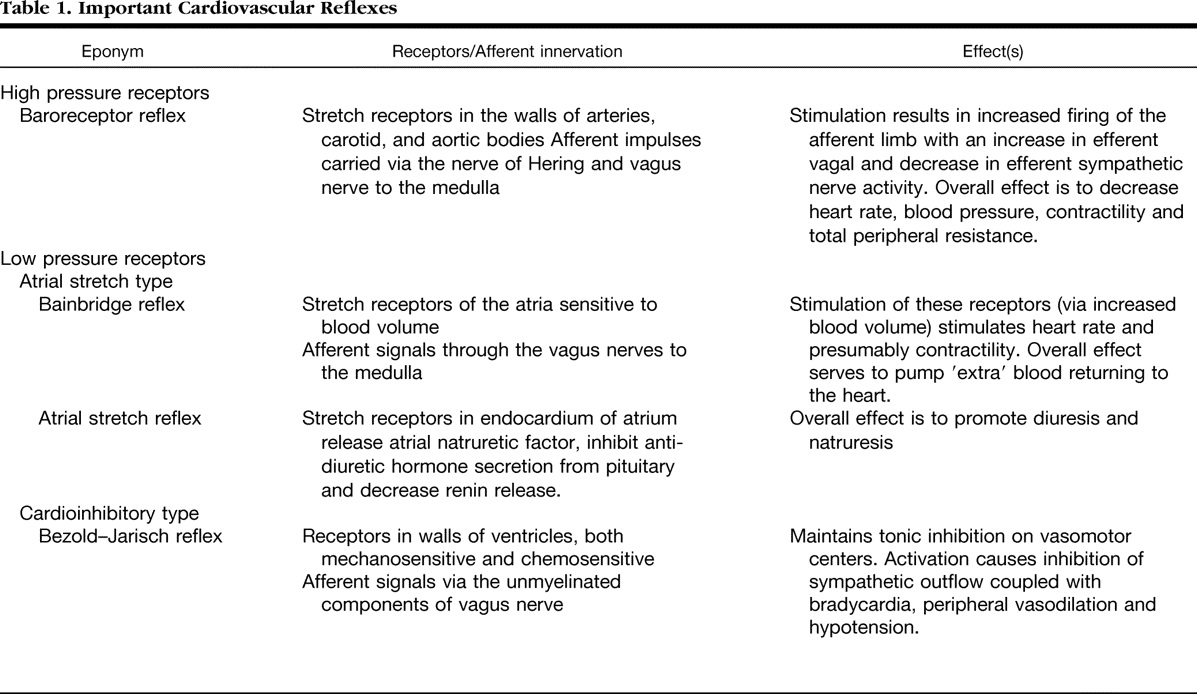
It is interesting to note many of these reflexes cause hypo-tension, bradycardia and hypopnea (Even near Apnea.) The word dyspnea is surprisingly not used .It is highly plausible many of the unexplained dyspnea we see in otherwise healthy population is attributed to acute or chronic volume overloading or under-loading of right heart.
Role of PFO in right heart dyspnea
PFO is a natural decompressing orifice in the IAS guarded by a flip-flap safety valve which is a remnant of septum primum .Though it can flow either way , since the flap of the valve is larger in LA side, it gets closed when LA pressure raises but opens up , if RA pressure raises making it more often a right to left shunt at times of elevated RA mean pressure. In isolated right heat pathology , this communication shunts right to left and adds a new dimension to cardiac dyspnea (Now, It becomes a hypoxic /biochemical dyspnea over and above the right heart stretch related dyspnea )
Other mechanisms in right heart dyspnea
Pulmonary arterial stretch and altered QP : Role of ventilation perfusion mismatch should also be considered as a cause for dyspnea in isolated RV pathology. The term V/Q mismatch is a poorly understood term fro me. My Inference is, since RV contraction provides the Q in the equation V/Q .Whenever Q falls V has to fall to maintain neutrality causing net hypoxia and dyspnea.
Final message
Dear fellows, never hesitate to attribute the origin of dyspnea, to elevated RA mean pressure /RVEDP. It is due to RA/RV stretch secondary to volume and pressure overloading with a perfectly normal pulmonary capillary wedge pressure or LVEDP. As in the left heart ,this occurs both in pathological as well as perfectly exaggerated physiological times.
Reference
1.Bainbridge FA. The influence of venous filling upon the rate of the heart. J Physiol. 1915 Dec 24;50(2):65–84. [PMC free article] [PubMed] [Google Scholar]
2..A J Crisp, R Hainsworth, and S M Tutt The absence of cardiovascular and respiratory responses to changes in right ventricular pressure in anaesthetized dogs. J Physiol. 1988 Dec; 407: 1–13(This paper actually undermines the importance of RV receptors. It is still perplexing to note both the inflow into RV (ie RA and the out flow pulmonary artery circuit has richly innervated by receptors , its difficult to accept why we have failed to get much evidence for RV stretch receptors) Its potentially great area of research for cardiac physiologists. That will be a tribute to the greats like Bain Bridge and Bazolds Jarich.)
Posted in Uncategorized, tagged his bundle capture in vt, narrow qrs vt, SVT VT with abberrency, vt entry and exit point, what is narrow qrs vt, what is wide qrs vt, wide qrs vt on December 4, 2019|
Why VTs have wide QRS complex?
Brief answer: VT usually presents with wide QRS tachycardia because it originates in ventricular myocardium, travels muscle to muscle instead of the normal conduction system. However, VTs need not be wide always, if it captures the conduction system early and more proximally it can be as narrow as SVT.
Further reading: Only for cardiology fellows
Two empirical statements are made here. (The scientific chances of both being reasonably correct are fair)
It’s obvious, not all VTs are dramatically wide. When it is not wide, they test our knowledge and patience. Let us be clear about the factors that determine the QRS width during VT. Once we know this we can have our own inference.
What determines the width of QRS in VT?

1.Origin of VT
The focus of origin is extremely important. Pure myocardial focus distal to the conduction system is invariably very wide. We know VTs originating right over the fascicles are narrow.
2.His Purkinje breakthrough
The time taken to capture the normal septal conduction system is a critical determinant of QRS width during VT.This makes the VT from septal origin narrower.VT arising from the free walls obviously takes a longer time to engage the HIS Purkinje system. Imagine , If VT originates from the lateral mitral annulus, how much time it may take to reach RV free wall and lastly RVOT. Here the VT will become bizarrely wide.
3.The structural integrity of His Purkinje
It is important to emphasize a fact , even if the VT captures HIS Purkinje early, if they are diseased , still the VT will be wider.(Example bundle branch reentry in DCM in which VT keeps going around the conduction system still, it’s wider)
4.Course
Length of the re-entrant circuit. Macro reentry is expected to be wider. Focal or micro reentry will often be narrow, provided the distal circuit is not diseased.
5. Scars as barriers and boulders
If the VT circuit is interrupted by random scars en-route (from origin to exit) the VT width prolongs. (Evidence for scars is often visible in sinus rhythm ECG as notches /slurs or fragmentations in QRS )
6.Exit point of VT
This is a poorly understood term (at least for me) It is believed, VT can exit only epicardially. The line joining the focus of origin and the exit point is expected to decide the QRS axis. The problem comes when VT breaks out multiple paths and possibly sub-endocardial as well.
7.LV dysfunction
A severely dysfunctional ventricle can stretch the QRS irrespective of conduction system integrity.
8.The Ionic milieu of cells Interstitial resistance
We know, biological current is nothing but Ions in motion. So, no surprise it can alter the QRS morphology. The classical example is hyperkalemia , that can make ECG a wide and blunt sine wave. Local acidosis, hypoxia also influence the QRS duration.
9.Drugs
Any drug which has class 1C or 3 properties can slow the VT circuit velocity. Typically flecainide is well known to make QRS wider. Amiodarone may reduce the ventricular rate. in VT instead of reverting it. Apart from this these drugs depress the ventricular myocardium severely and prolong the QRS width independent to its action on the conduction system.
10.Mechanism of changing width
VTs can have varying QRS width as reentrant circuits change or experience slow conduction due to autonomic influences. VT with downstream aberrancy is also possible as the VT rate by itself influences the conduction property distally.(Just lie SVT with aberrancy)
A paradox about the width of QRS in VT
A curious phenomenon is often seen, when VT occurs in patients with baseline ECG which is already wide (As in an ischemic dilated cardiomyopathy with LBBB/RBBB). Here, the VT prematurely stimulates viable muscles distal to the diseased HIS Purkinje system (Which they are deprived of early activation of till then) .They seem to relish the early arrival of electrical impulse by brisk activation that converts wide QRS complex to narrow one. (This behavior is one of the principles of cardiac resynchronization therapy where we attempt to rewire the heart with multiple leads and shrink the QRS.)
*One more mechanism of wide QRS sinus rhythm becoming narrow during VT is due to a concept called source -sink relationship. The VT delivers enough energy overcoming His Purkinje resistance downstream. (This property is used in HIS bundle pacing )
Postamble
*Forget about wide vs narrow QRS debate. A significant chunk of VTs falls within intermediate width QRS(100-120ms) . Whether to label these as wide or narrow QRS squarely lies on whims of the reader. (Should we take the widest QRS in 12 lead ECG? Pre-cardial vs limb lead etc are not clear) Unfortunately, we don’t have a separate algorithm for this category. This issue demands a separate discussion.
Posted in Uncategorized, tagged COURAGE ORBITA ISCHEMIA, post ptca care, progression of native vessel disease after ptca on November 24, 2019|
Whenever a patient is getting discharged after a PCI, the treating cardiologist often faces this situation.
So, you fixed the block in my coronary artery doctor. Thank you so much. Now, I can have a peaceful life, free from future heart problems. “Am I right doctor”?
I wish I can answer “Yes” to your query but I can’t for the following reasons.
I have fixed only a lesion that caused maximum obstruction. Atherosclerosis is a diffuse disease and you have minor plaques scattered across your coronary artery. These can grow at its own will. So you carry a definite risk remote from the current problem. (Don’t get frightened, read further, you have definite solutions to reduce this risk.)
How common is the progression of native vessel disease?
It varies from 10 to 40%. Mind you, the exact incidence directly depends upon the compliance of medical management, risk factor reduction, and adaptation to a new life healthy lifestyle. In effect, you (the patients) decide the incidence.
One surprise phenomenon (though unproven) might happen. Since the tightest lesion is jailed with a scaffold the minor lesion is preselected to an accelerated process of atherosclerosis if medical treatment is not properly followed.
Dr.Zellweger from the university hospital, Basel, did an extraordinary study with 400 patients, meticulous 5 years follow up with SPECT and found remote lesions accounted for 40% of future events (Basel Stent Kosten-Effektivitäts Trial [BASKET]) The other study by Glazer and concurred with this. These studies reiterate the importance of taking care of the entire coronary artery instead of focused piecemeal care by scaffolds.
Does a proximal DES protect a distal lesion in the same artery by the drug effect?
It is a good thing to happen at least on paper. A proximal LAD with the latest generation Everolimus coated stent is expected to keep the distal LAD drugged for few months at leas.( with anti-mitotic activity) Thus preventing the progression of distal lesions.
No, I can’t believe this.In this era of momentary touch on sidewalls of artery by drug-eluting balloon (DEB) shown to do wonders, anything is feasible. Chacko’s (Ref 2 : JACC CV Interventions 2009)observation has a possible answer for this. It showed BMS vs DES didn’t make any difference in remote lesion progression.
Final message
These studies reaffirm one vital truth. Stents are temporary solutions to a permanent, systemic disease of the vascular system .Stents are indeed a major revolution in CAD, “if and only if” it’s used in a highly selected CAD population. Global attempts to project cath labs as a tool to control human atherosclerosis is a typical example of flawed science. The only effective way to tackle this menace is to faithfully follow overall healthy living, assisted by drugs.
This is the Editorial in response to Zellweger’s article

Reference

Postamble
One of my patients asked some time ago. If stents are the definite remedy for severe arterial narrowing, why not stent all my lesions (even the minor ones ) prophylactically doctor, so that it will not become tight at a later date?
That’s a good query. Your doubt is genuine , appear logical as well. But, unfortunately, it will be the most dangerous thing to do*. Metals are never friendly with the coronary arterial wall. We should use it extremely judiciously and only with tight flow-limiting lesions. These metals require annual (rather permanent) maintenance. Its taken care by multiple antiplatelet drugs. If for some reason your maintenance is erratic or the drugs fail to act you are at more risk of a future event.
(* This is what has happened (happening) in the past, that demanded urgent publication of appropriate usage criteria)
Now, the current belief among the “fair thinking cardiology community” is dramatically changing. It’s leaning towards non-stent management even with significant flow-limiting obstructions in otherwise stable patients(SIHD). This belief is accruing more and more evidence base (The COURAGE 15 year follow up / ORBITA/ISCHEMIA) All these studies confirm the emerging doctrine and bring back some semblance of sense into the cardiology community.
Posted in Uncategorized, tagged ecmo north south syndrome, harlequin syndrome, va ecmo side effects on November 12, 2019|
Differential cyanosis classically occurs in PDA with reversal of shunt when raised PA pressures /PVR is able to supersede the systemic Aortic pressure and drive the blood from LPA to descending Aorta bringing down the lower limb saturation.
Of course, this can be undone by the presence of any other intra-cardiac shunts or aberrant left subclavian that arising from the desaturated descending aorta.
Other causes of reversed differential cyanosis
Where the upper body is cyanosed (desaturated) and the lower half is not. There is a conventional list of conditions.
(*This occurs due to streaming effect ) Highly saturated superior vena cava (SVC) blood into the right ventricle, reach MPA / through a PDA, and to the descending aorta, with streaming of more desaturated blood from the inferior vena cava (IVC) into the LA through PFO (Ref Yap S H Pediatr Cardiol. 2009 )
Now let us add one more cause for reversed differential cyanosis in the Modern Era
It is seen with ECMO in VA connection (Often reported in babies ) . The Aorta has high oxygen content entering from the femoral cannula going up into the Aortic arch., while deoxygenated blood from LV (because of failing lungs) reach antegradely to the Aorta. Ideally, the ECMO is expected to supply the entire aortic arch and hence oxygenation is uniform all over the body. It rarely happens as some amount of flow will come from LV unless its in asystole. However, If the severely dysfunctional heart tends to recover & lung oxygenation is very poor as well, the LV stroke volume competes with highly oxygenated blood coming from below ( femoral inflow ) into the Aorta , creating a watershed zone . This makes the deoxygenated blood perfusing upper half of the body and hyper oxygen saturation lower half. This is been referred to as North-south syndrome or (Harlequin syndrome the famous Italian comical character)

How to manage North-South syndrome?
Reference

Posted in post dilatation, Tips and tricks in cath lab, Uncategorized, tagged balloon inflation time in ptca, how to avoid post dilatation, malapposition and balloon inflation time, tips and tricks in ptca on November 1, 2019|
I asked some of my experienced colleagues, how much time they inflate the balloon to deliver a stent? Most answers were spontaneous and unanimous “It’s hardly 10 seconds, few said maybe up to 15s.
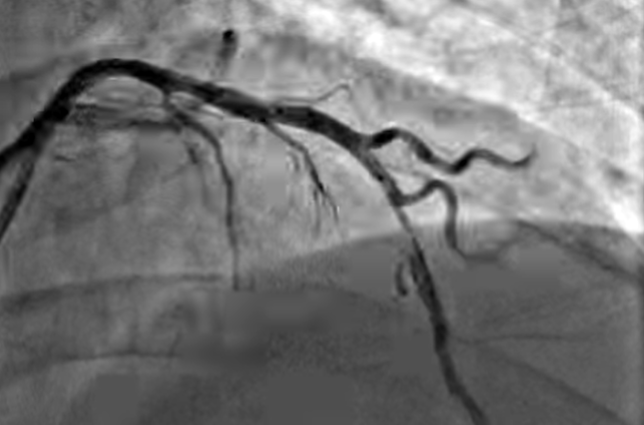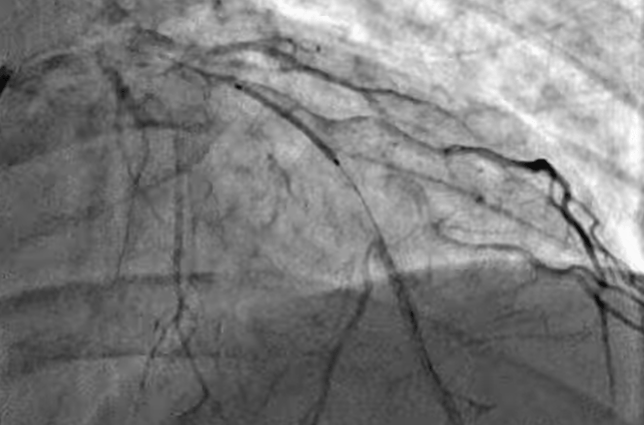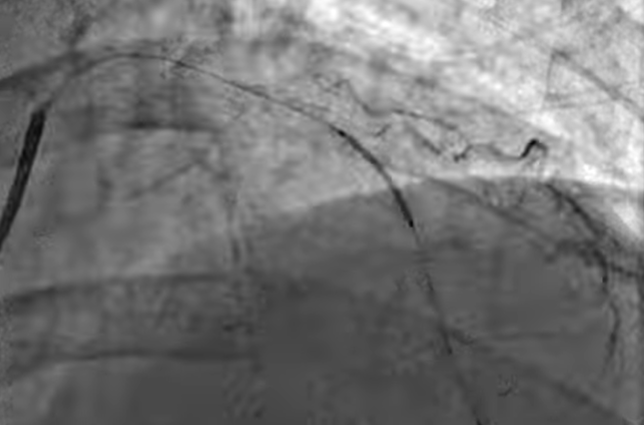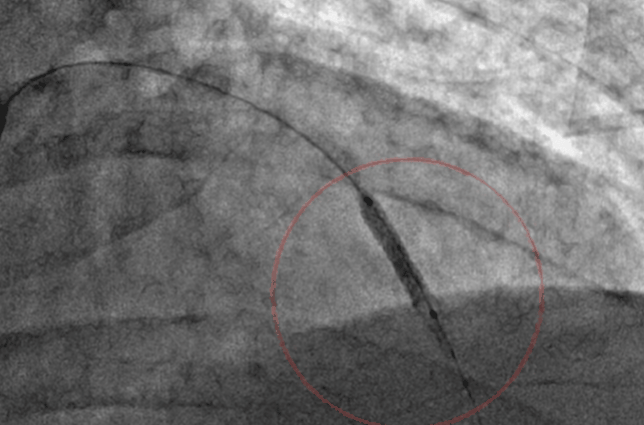
Can prolonged balloon inflation time reduce the need for post dilatation and prevent mal-apposition?
We know high-pressure Inflation( up to 20 atmospheres ) was a big revelation in the science of PTCA more than two decades ago. (Antonio Colombo JACC 1995 ) He proposed and proved high-pressure inflation eliminated the need for routine anticoagulation following stenting as approximation was better. He also pioneered the concept of dual antiplatelet therapy (DAPT) in the PCI arena.
Similarly, prolonged balloon Inflation (30 to 60sec) could be another trendsetting tip to prevent malposition. It delivers more sustained pressure. Its believed the imparted centrifugal force and the inbuilt radial forces add up to the stent vessel wall Interface and prevent mal-apposition.
Is there a downside to high-pressure Inflation?
There must be few. Potential new Ischemic events and arrhythmia. In calcium laden plaques( spur) risk of perforation may be enhanced.
Final message
I don’t know why this concept never took off. Many of us still fear to inflate the stent balloon no longer than 10 to 20 seconds? Adhoc post dilatation with short NC balloon appears mandatory in areas of mal-apposition. Meanwhile, we also understand sustained (30-60s) high-pressure initial inflation could deliver the stent in a more synchronized and smooth fashion with a perfect metal/vessel wall interface. Further , prolonged balloon inflation times could make a routine (By the way who does routine ?) IVUS/OCT redundant.
What do the experts say? What does science say? There is one meta-analysis that clearly says the advantage of long inflation time. This issue becomes much more relevant as it could avoid post dilatation which all of us know can be tricky. In fact, every balloon dilatation should be technically counted as another PTCA procedure and adds up to net total risk.
Reference
1.M. Saad, M. Bavineni, B. F. Uretsky, and S. Vallurupalli, “Improved stent expansion with prolonged compared with short balloon inflation: a meta-analysis,” Catheterization and Cardiovascular Interventions, vol. 92, pp. 873–880, 2018. View at Google Scholar
Posted in Uncategorized, tagged acc aha esc steminstemi ccs management guidelines, acute coroanry syndrome managment, decision making in pci, evidence based coronary care, fame 1 fame 2, ffr cut off value, ffr vs oct, forza trial study tct 2019, nstemi unstable angina, vulnerable plaque on October 2, 2019|
Rules of the PCI game
So what should we do in a case of 70 % LAD with .9 FFR ? (Still shabby looking, eccentric plaque, looks vulnerable with a thin cap on OCT)
Which is correct?
All can be fair depending upon the clinical scenario.
In the ACS setting, one can’t afford to ignore these lessons.
Many would argue even in CCS setting it need to be tackled with PCI.
But isn’t also a fact, (maybe, we have been taught wrong as well ) non-flow-limiting lesions are more at risk in terms of ACS risk.
Hmm . . . then why we Insist to celebrate the concept of FFR and its magic cut off of .75?
Do we practice coronary care at its height of confusing times ? or Am I make it appear so?
Watch this, (https://rutherfordmedicine.com/videos )It might help you to get a better answer. Its called FORZA study. freshly delivered at TCT 2019, San Francisco.It compares FFR vs OCT guided PCI

Posted in Criteria and Nomenclature, ischemic cardiomyopathy, Uncategorized, tagged defintion of ischemic cardiomyopathy, MOGES classification, what is ischemic cardiomyopathy, who whf classification of cardiomyopathy on September 21, 2019|
The term Ischemic cardiomyopathy(ICM) was originally coined by Dr. Burch from Tulane University, New Orleans, USA in 1970. For many decades there was skepticism regarding the existence of such entity. WHO classification over the years never included this term. ESC working group of 2008 (Elliott P, European Heart 29(2):270–276) decided not to include CAD as a cause for cardiomyopathy. Even the current MOGES system doesn’t invoke CAD as a cause for cardiomyopathy. But, I am sure, most of practicing cardiologists would agree, there is a need for such an entity.
Why there is much reluctance to diagnose Ischemic cardiomyopathy as a distinct entity?
It is because of the basic principle, that cardiomyopathy should be a primary disease of cardiac muscle. (or at least secondary ).The presumption is, Ischemia per se doesn’t lead to muscle disease as such. It is just nutrition deprival.
Does this justify?
No, not at all. When a cardiac muscle is chronically deprived of nutrients it goes for necrosis, dilatation, scarring and dilatation, and progressive LV dysfunction. At some stage, it becomes true muscle disease or its equivalent (Secondary cardiomyopathy).In fact, adverse remodeling, Infarct expansion, extension lead to myocyte disarray, slippage and apoptosis, and cellular and interstitial fibrosis. All these changes are similar to Idiopathic (Postmyocarditis)cardiomyopathy.
What happens in the real world?
Even though there was some hesitation to diagnose ICM in the past, gradually the term shrugged of its taboo in academic circles. In heart failure clinics the only question seems to matter for everyone is, Is it Ischemic or non-ischemic DCM? Surgeons and EP guys also actively pursued the term Ischemic cardiomyopathy while they are selecting patients for CABG or CRT/ICD etc.
Further, in the research world involving community-based heart failure cohort, they required a basic distinction between the group of Ischemic from Non-Ischemic cardiac failures.
DUKE university definition (By Felker et al)
I think DUKE ended the controversy in the Nomenclature of Ischemic cardiomyopathy. It suggested the following to diagnose ICM (Read REF 2)
* We are analyzing our data (Madras medical college, Chennai India) and propose to write WHO/WHF to include the following additional criteria to diagnose ICM.
4. At least 6 months should be elapsed between the MI and diagnosing Ischemic cardiomyopathy,
5. Must have significant LV dilatation & global Hypokinesia(With or without regional variation).
6. At least one episode of clinical heart failure is required before labeling it as Ischemic cardiomyopathy.
Other definitions that endorsed Duke
STITCH criteria *Surgical therapy in ischemic DCM study ICM was defined CAD with cut off EF < 35% with triple or double vessel disease.
iFAQs in Ischemic cardiomyopathy
1. Can we diagnose ICM without a history of MI?
This is tricky. As we all aware its very much possible as in silent MI of diabetes. One more possibility is even chronic coronary syndrome with microvascular dysfunction can lead to ICM.
2. Can Ischemic cardiomyopathy present as HFpEF or RCM?
While most Ischemic Cardiomyopathy present as DCMs with HFrEF, It is currently not clear how much of Ischemic heart failure present as HFpEF and if so they can’t be included technically as Ischemic cardiomyopathy in spite of the fact they present as HF.(as EF would be >50%)
3.When does a Post MI failure become Ischemic cardiomyopathy?
If the definition of STITCH or DUKE is applied, any acute STEMI can fulfill criteria of ICM. Hence it advisable to have a time limit say 6 months following MI to be referred to as Ischemic DCM. Pathologically to call it true cardiomyopathy, scarring, dilatation is required. Myocytes should be in independent self-destruction mode irrespective if hemodynamic conditions.
Response to treatment
The only purpose to diagnose ICM is to try to remove the I from ICM( ie Ischemia) Unfortunately, it is not an easy task. (While correcting Ischemia in ACS seems to be such an easy job.)
Following principles apply.
Final message
I agree, many times our valuable time is wasted in renaming /Altering /relabeling a disease /process or pathology without any useful purpose. Medical nomenclature and classifications are done to make diagnosis simpler, choose an appropriate therapeutic modality and make a positive impact on the outcome.
In that sense, segregating ICM from other causes of cardiac failure do help in choosing a specific management strategy.
Let us welcome MOGES, It is the most comprehensive cardiomyopathy classification system (Like TNM classification for cancer). Still, I am not clear why it hasn’t included CAD in that system. Thanks to Dr. Burch who thought of this 50years ago.
Reference
1.Burch, G. E., Giles, T. D., & Colcolough, H. L. (1970). Ischemic cardiomyopathy. American Heart Journal, 79(3), 291–292.
2.Felker G.M, Shaw L.K, O’Connor C.M (2002) A standardized definition of ischemic cardiomyopathy for use in clinical research. J Am Coll Cardiol 39:210–218

History of cardiomyopathy classification
The landmark thoughts originated in 1972 .When Goodwin and Oakley defined cardiomyopathies as the heart muscle diseases of unknown cause and described them as dilated (DCM), hypertrophic (HCM), and restrictive (or obliterative) (RCM) cardiomyopathy types.
WHO adopted it mostly and suggested Primary and Secondary cardiomyopathy in 1980. In 1995 WHO revised it.
The current MOGES classification doesn’t mention about Ischemic etiology
Posted in Uncategorized, tagged master health check up, medical ethics on September 17, 2019|
Master health checks* , superficially look like a perfect modality to practice the greatest medical concept ie “Prevention is better then cure” .Let us detect all human diseases early , prevent its progression, regress it or completely cure it . Absolute bliss is it not?
Why then articles such as this one should ever get published, that too in one of the prestigious journal of medicine?
*Master health check .( Also referred to as annual General health checks.)

While the title itself is provocative, it adds a tag line which is still more a shocker.
There are specific well-researched reasons for this preventive health check fiasco.The masters, who were originally the guardians of health soon became disease mongers.In the process, the primary aim of propagating the doctrine of “prevention is better than cure”, could not reach its desired goals. Instead of ignoring and reassuring the minor deviation of biological data and Imagery generated, they became a perfect feed for the hunters who are after the trivial and non-existing illness.
Final message
Good intentioned health checks are always welcome in selected high-risk population say pregnant women/children of developing a world (As in endemic countries of rheumatic fever) Also cancer , CAD , screening in people with a positive family history can be critical.
However, when these masters of health deviated and started making a living out of apparently healthy people. ( The side effects reached monstrous proportions hiking global health cost in a meaningless way).People, especially in counties with poor resources, are the ultimate sufferers, as the cost and efforts are diverted, to fix the health of healthy, while people with true illness continue to struggle.
Will the WHO* wake up and intervene against this skewed practice of routine master health checks in healthy, that are rampant in both rich and poor countries. Ideally, doctors should order preventive health assessment for those who may need it.
There are enough grounds for public Initiated periodic walk-in health checks to be banned (or at least restricted)
*WHO is world health organization




