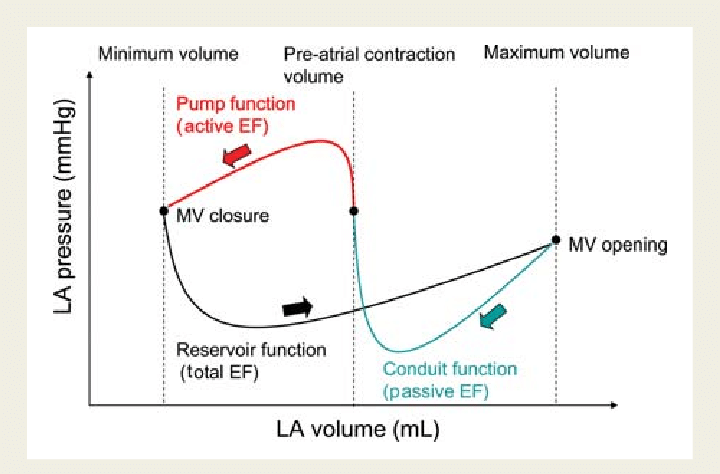
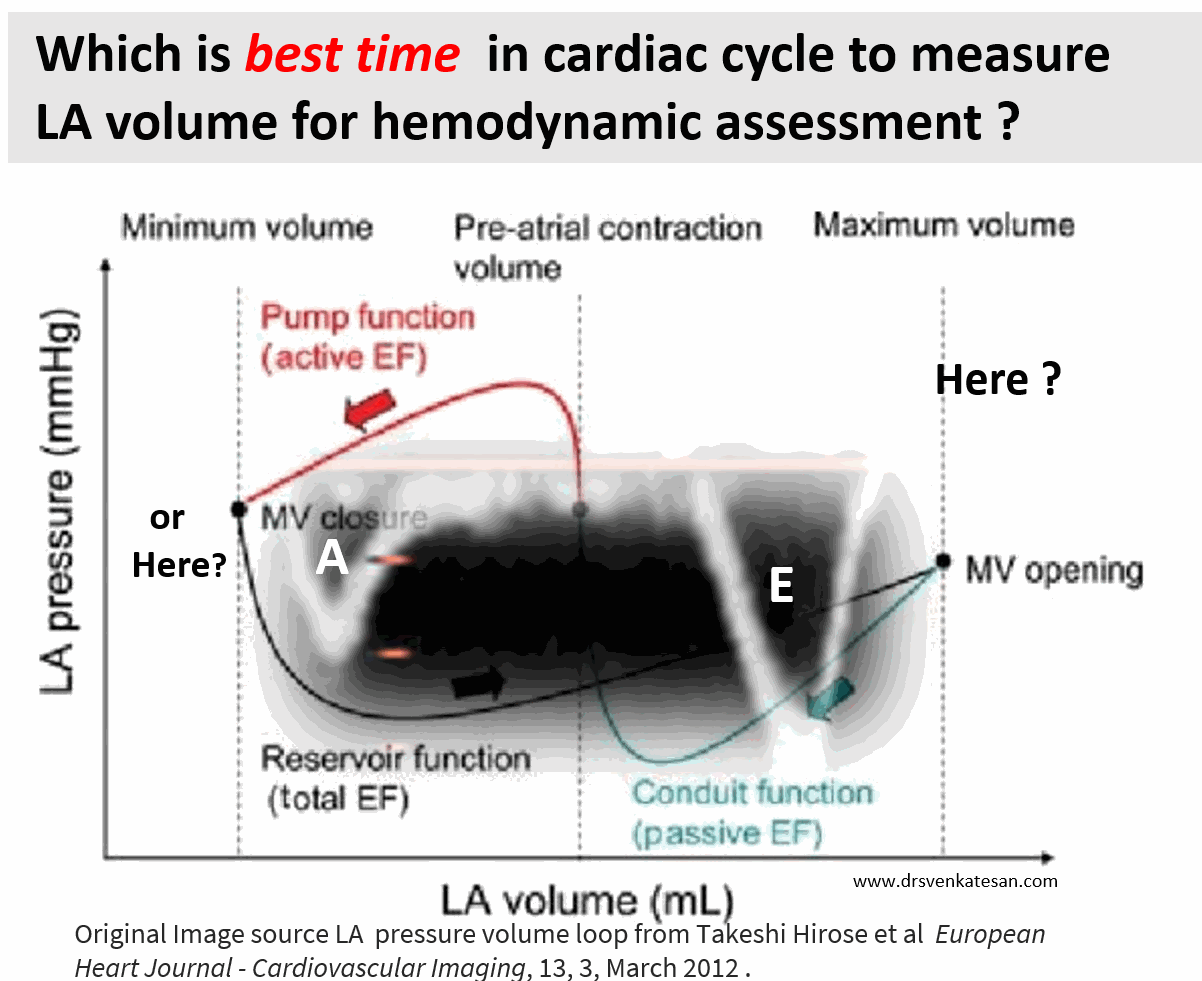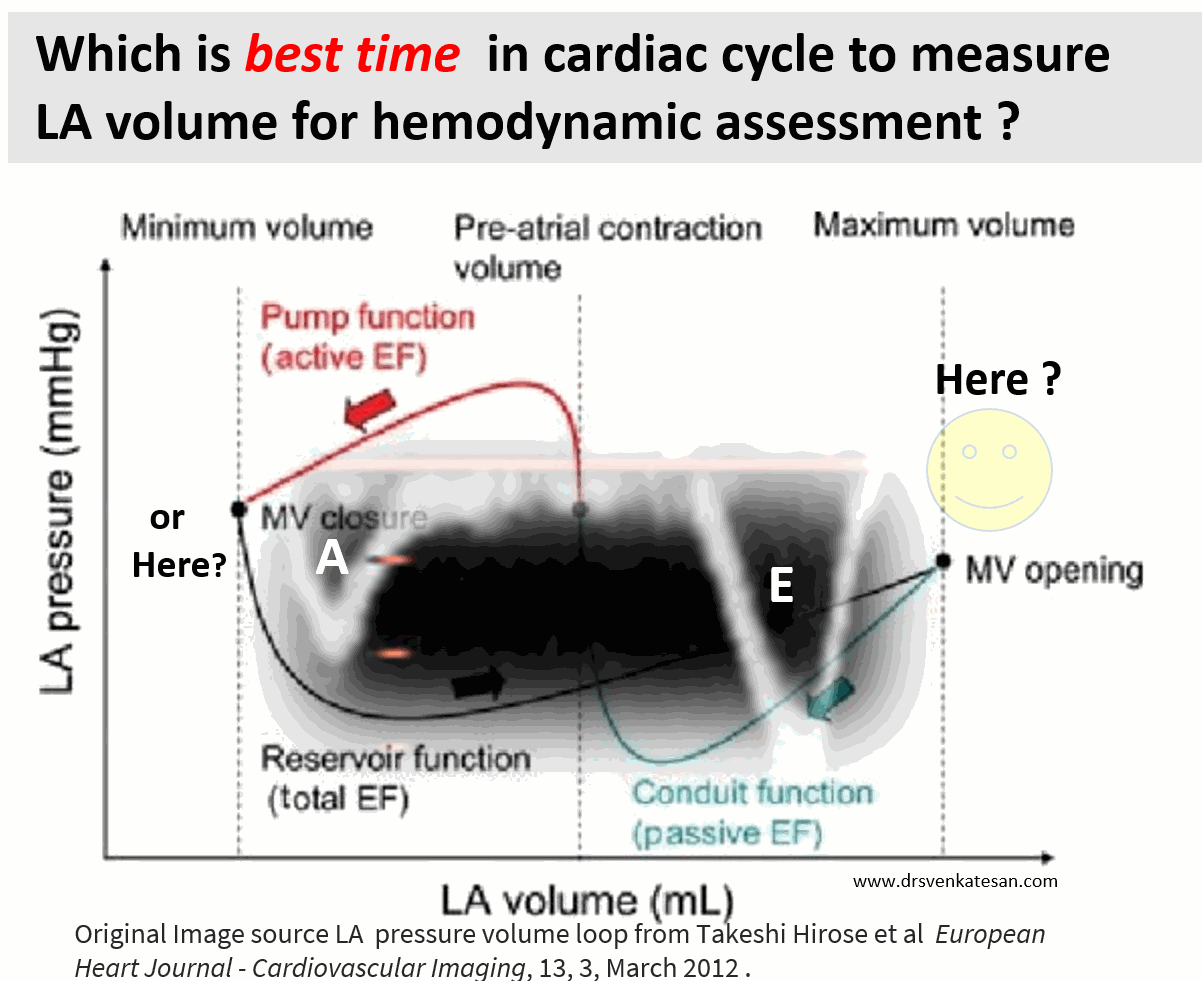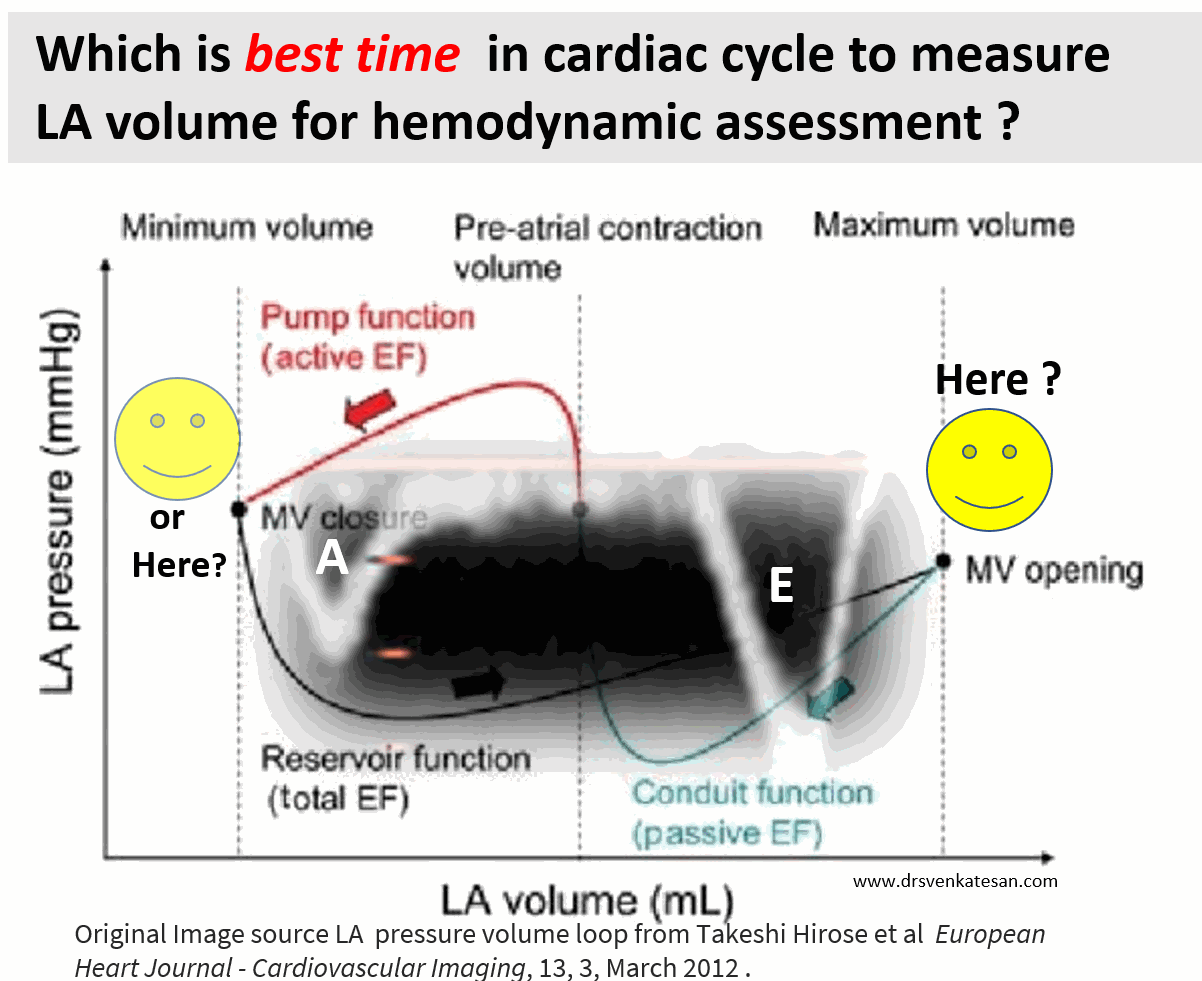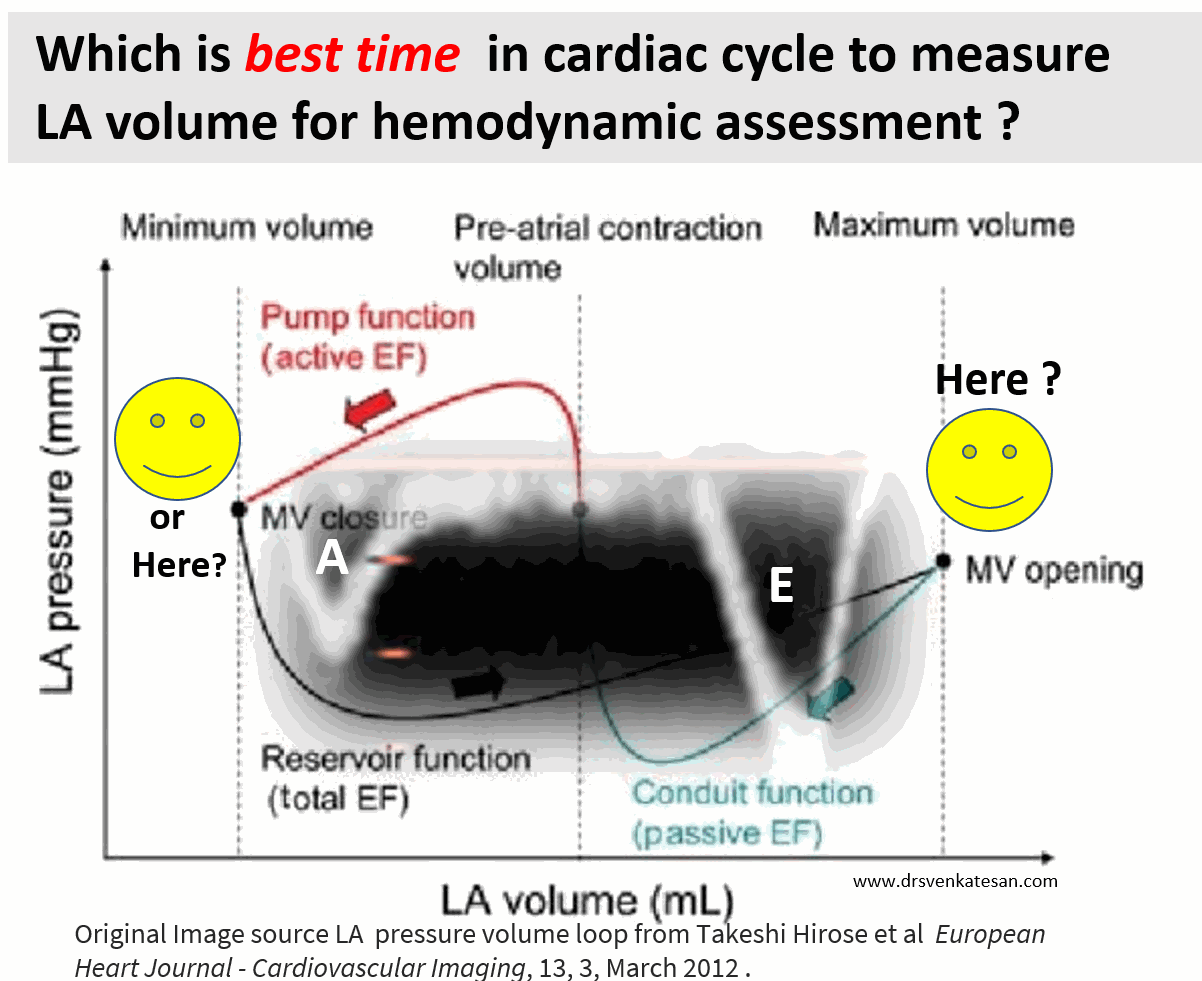
Cardiologists have been trying for the last two decades to prove PCI is superior or at least equal, to CABG in multivessel CAD. We desperately needed studies to prevail over FREEDOM and SYNTAX which favored CABG.
FAME series , though never had an intention to compare PCI vs CABG , now we have used the platform to upend it to take on the CABG in multivessel CAD. ( FAME 3)
FAME 1 (Fractional Flow Reserve Versus Angiography for Multivessel Evaluation)
Purpose
The FAME 1 study aimed to compare the efficacy of FFR-guided PCI versus angiography-guided PCI in patients with multivessel coronary artery disease (CAD). The goal was to determine whether using FFR to identify functionally significant stenoses (FFR ≤ 0.80) for stenting, rather than relying solely on angiographic appearance.
Inference
Established that FFR-guided PCI is superior to angiography-guided PCI in multivessel CAD, reducing unnecessary revascularizations and improving outcomes.
FAME 2
Purpose
FAME 2 sought to evaluate whether FFR-guided PCI plus optimal medical therapy (OMT) was superior to OMT alone in patients with stable CAD and at least one functionally significant stenosis (FFR ≤ 0.80).
The study concluded that FFR-guided PCI is beneficial in stable CAD when ischemia is present, reducing the need for subsequent urgent revascularizations compared to OMT alone, though it did not significantly reduce rates of death or MI
FAME 3
Purpose
FAME 3 aimed to determine whether FFR-guided PCI using contemporary drug-eluting stents was non-inferior to CABG in patients with three-vessel CAD. The study sought to compare these two revascularization strategies in terms of clinical outcomes, testing the hypothesis that FFR-guided PCI could achieve similar results to CABG by targeting only functionally significant lesions.
Conclusion
At 1 year, FFR-guided PCI did not meet the criterion for non-inferiority compared to CABG for the primary endpoint of MACE (death, MI, stroke, or repeat revascularization), with event rates of 10.6% for PCI vs. 6.9% for CABG.
5 years later in 2025
FAME 3 : 5 year follow up data , released in 2025, tries to confirm the non inferiorty of PCI over CABG in a larger sense.

The study concluded that in patients with three-vessel CAD, FFR-guided PCI is not non-inferior to CABG, with CABG remaining superior for reducing MI and repeat revascularization. However, FFR guidance still refined PCI by limiting interventions to functionally significant lesions.
Did the FAME 3 study compare FFR guided CABG vs FFR guided PCI ?
No, none of the FAME studies (FAME 1, FAME 2, or FAME 3) directly compared FFR-guided CABG versus FFR-guided PCI. Each study had a distinct focus involving FFR-guided PCI, but none incorporated FFR guidance into CABG as a primary comparator. Here’s why this comparison could be meaningless.
Final message
Truths express themselves. We can’t force it to happen.
Reference