Is “Non-flow limiting“ coronary lesions more prone for ACS ?
- If your answer is “No”, you can skip this article.
- If your answer is “Yes” , you need to read this article.
ACS is the commonest cardiac emergency .Thousands of patients are treated every day.Millions of dollars are spent.Bulk of the cardiologist’s life revolves around this entity.
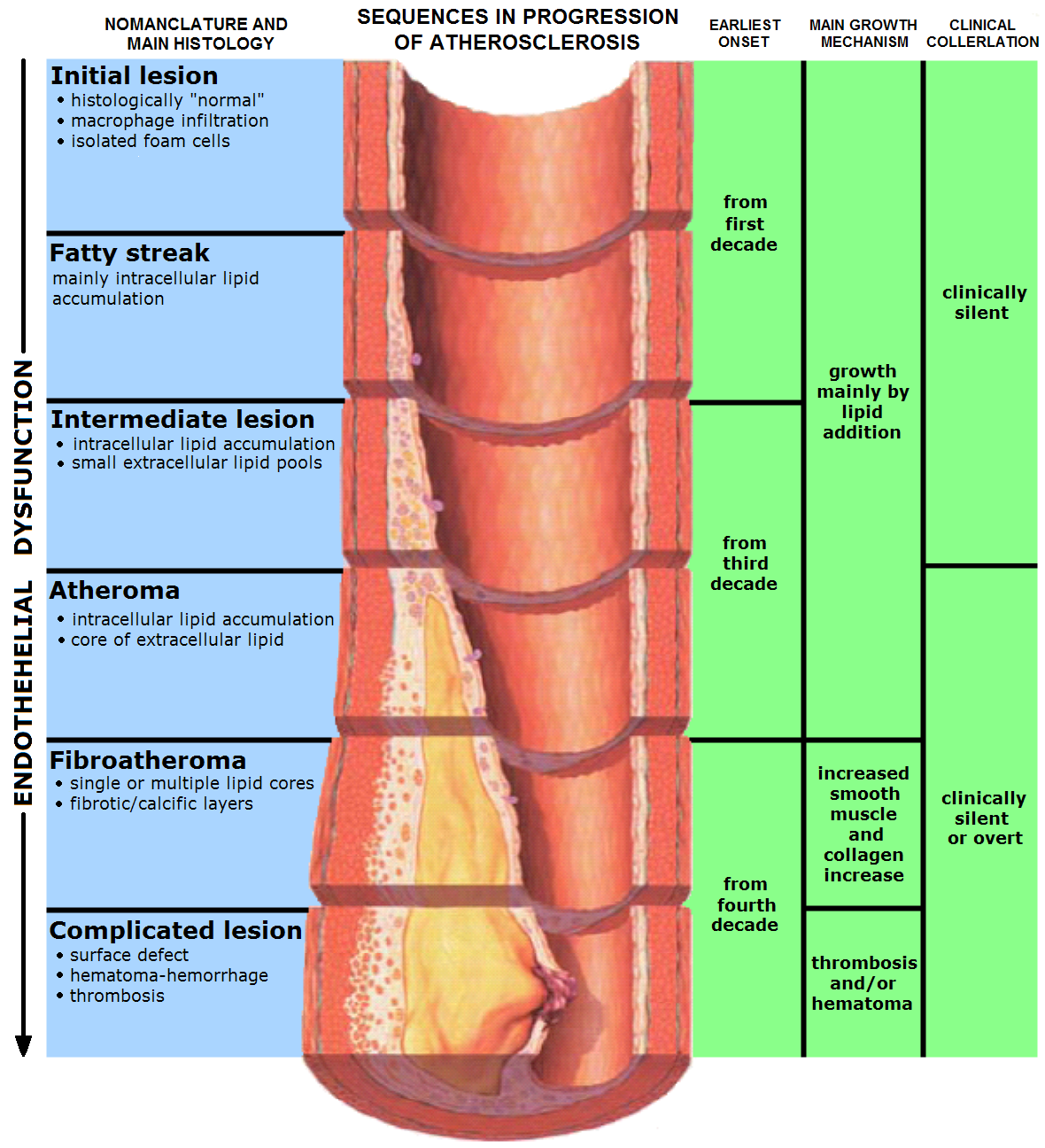
Scattered atherosclerotic plaques in coronary artery lead to ACS either in a random fashion or in a predictable manner .
Still, we are highly uncertain about which lesions are likely to result in ACS ! Some time in the beginning of 21st century, the main stream cardiology media were abuzz with the concept, that non obstructive , non-flow limiting lesions are more prone for ACS rather than more tight stenosis.
This reasoning was based on few studies, that lacked solid scientific proof . In fact the initial observation was not made in living coronary arteries rather by autopsy observations .(Later live virtual histological studies came , but didn’t confirm this !)
Surprisingly the degree of anatomical narrowing was conferred vulnerability , when we know plaque compositions , morphology and hemo-rheological factors are many fold important in precipitating ACS . (Lipid content , fibrin cap thickness, eccentricity , etc)
So where is the truth hidden?
Is it really possible, lesser the stenosis more is the propensity for rupture ?
We need to introspect .
“In all probability, it is a meager statistical illusion”
For every tight lesion there are as many minor lesions scattered around in a given a coronary artery. These can progress into ACS later.
It is basically wrong to assume non-flow limiting lesions are more prone for ACS than non-flow limiting lesions.To believe so , seriously underestimates the culpability of big lesions .It appears a coronary mockery to me !
At best , we can conclude non-flow limiting lesions are not benign and can be an important source of ACS.
An unscientific chain reaction !
If we start believing non flow limiting (say 30% stenosis ) is more prone for ACS , why we are not stenting all those lesions ?
If the above concept is is applied in cath lab routinely , the principle of FFR which relies solely on hemodynamic impact will crash into the dustbin !
Some more truths
However , It is indeed true when a plaque is hardened by severe sclerotic process or calcification it is less prone for rupture and clinical ACS but can be a source for stable angina.
Is it justified to assume , larger the plaque the harder would be it’s content that resists ACS ?
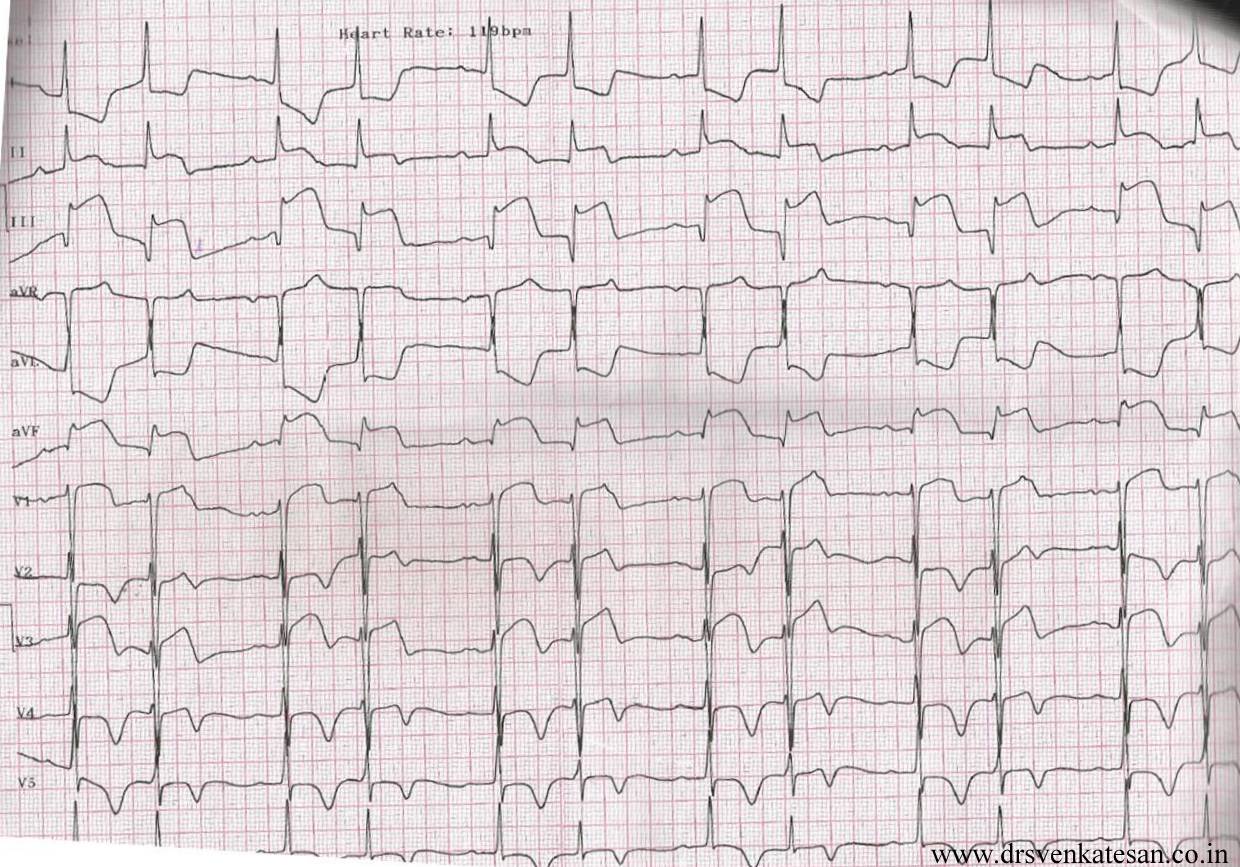
Meanwhile , we also know there need not be any lesion at all to cause an ACS.( In a young smoker , 100 % thrombotic STEMI is possible over an area of coronary erosion caused by endothelial dysfunction ! So , where do we go from here !)
Let us be clear
Are you confused more ! . . . after reading this article, let us clear it by two-line summary !
As on 2014 ,
- Symptomatic flow limiting lesion are tackled by stents.
- All non-flow limiting lesions are treated by high dose Statins and vigorous medical management.
Final message
Contrary to popular perception, tight lesions are more complex, eccentric , soft and are at immediate risk of ACS.
Non flow limiting lesions remain static in most, regress in many , still carries distinct risk of progression into full blown ACS , at any time if conditions are favorable.
Fixed concepts and ideas in medical science do not help us taking medicine forward. Especially so, when these are based on assumptions and approximations. If only we redo these studies with the currently available technology (FFR/OCT/NIR the conclusions would be dramatically different. !
Waiting for someone to nullify such false concepts in a more scientific way !
Reference
2.Glagov S, Weisenberg E, Zarins C, Stankunavicius R, Kolletis G. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987; 316: 371–375.
3.Fuster V, Lewis A. Conner Memorial Lecture. Mechanisms leading to myocardial infarction: insights from studies of vascular biology. Circulation 1994;90:2126-2146.
4.Ambrose JA, Weinrauch M. Thrombosis in ischemic heart disease. Arch Int Med 1996;156:1382-1394
Postamble
This post was written in 2014. Happy to find a scientific proof to this concept in 2018.
Source : PROSPECT study
Retrospective angiographic studies and the prospective PROSPECT (Providing Regional Observations to Study Predictors of Events in the Coronary Tree) study have shown that plaques with severe stenosis carry a higher per-plaque risk for producing clinical events than plaques that cause no or non severe stenosis.
However, such lesions are few, and overall, most ACS are precipitated by plaques without significant stenosis on an antecedent angiography
weeks or months before. This epidemiology is consistent with the distribution of TCFAs, as shown by a combined angiography and optical coherence tomographic imaging study of nonculprit lesions.
Lesions that caused severe stenosis were twice as likely to be
TCFAs than lesions with only non severe stenosis, but the total number of TCFAs with nonsevere stenosis was three times higher than those with severe stenosis. The mild pre-existent stenosis of most TCFAs and ruptured plaques is explained by expansive remodeling, because such lesions are, on average, large.
The long-held notion that mild to moderate obstructive coronary lesions are responsible for the majority of MIs has been challenged by studies that described significant narrowing in the days preceding MI. However, significant narrowing shortly before MI may be a result of (rather than a precursor) for rupture.
Read Full Post »








{kind=link}