William Heberden first introduced the term angina to the medical community in 1772. His descriptions became immortal. Still, no one would ever know what was the angina-related artery, Heberden was alluding to.
Now, some jobless cardiologist is asking this question after 200 years. How is angina from the LAD system differ from the RCA system? or let me put it another way, How does angina of anterior circulation (LAD) differ from posterior circulation (RCA/LCX)? Though there is distinct hemodynamic profiling of RCAvs LAD ACS, surprisingly, cardiology literature does not answer the chest pain aspect of it. One rare study, done 4 decades ago throws some light
Here is a curious little study, with a simple & crisp conclusion.

It concludes, that LAD angina rarely radiates to JAW or epigastrium. While RCA angina relay radiates to the left shoulder.
So, why does this happen?
What I could guess is the ubiquitous vagal fibers that travel in the posterior aspect of the heart, and carries pain signal directly up to the jaw whenever these areas become ischemia. LAD is less likely to irritate the vagus. Of course, there can be a definite overlap.
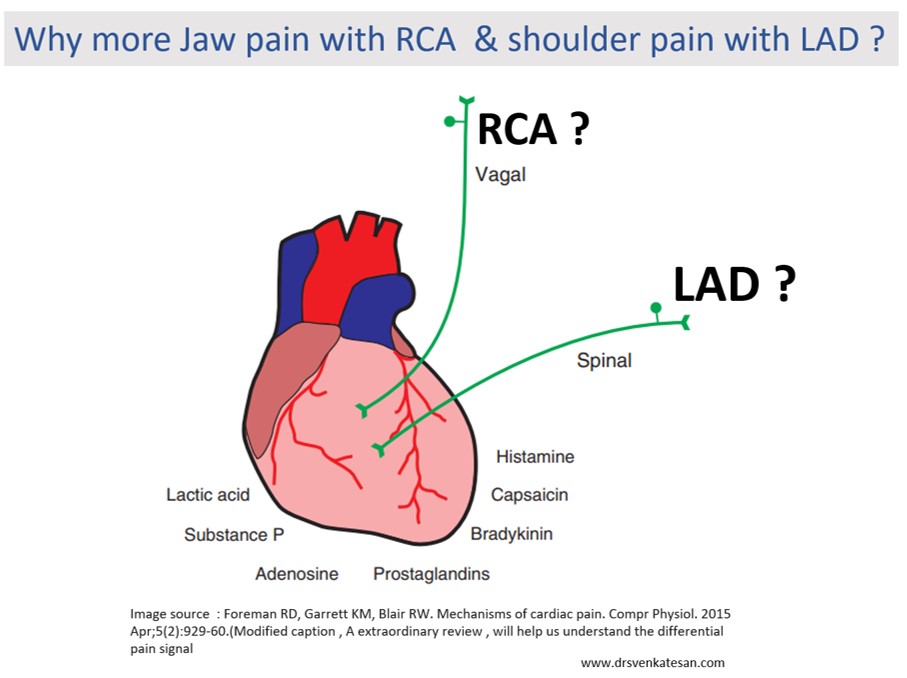
OMG, give me some time to keep in touch with basic science
Now, fellows of cardiology, please take a pause from your regular aggressive cardiac cath lab workouts and get a break at least once in a while. How does the ischemia of myocardial tissue generate pain? Why it is severe in some, trivial in others, and even dead silent in some,
The chest pain genesis is initiated by sensory electrical neural action potential, that captures the epicardial neural plexus first, switching over from somatic to the visceral pathway and trespassing the para ganglionic plexus and traveling further to the spinal cord. Where it may collide with other incoming sensory signals ascends in specific myelinated and non-myelinated neural cables, reaching the brainstem, interacting with local nuclei, and finally reflecting on subcortical and cortical pain matching centers. We haven’t yet located the exact center for anginal pain. (Perithalmic and amygdala could be closer to real centers)
So, it is a really complex sensory world yet to be understood fully. Mind you, I haven’t touched upon the neurophysics of referred pain, linked or clandestine angina.
- What is the effect of cardiac denervation, autonomic neuropathy, or on the perception of chest pain(Does a quadriplegic feel angina ? or post-transplant heart immune to angina ? (Gallego Page JC,Rev Esp Cardiol. 2001)
- Is it biochemical or neural, can substance P in blood cause pain hitting the amygdala?
- Will hypoglycemia and anemia cause angina due to lack of glucose and oxygen?
- Finally, how is Infarct pain is different from ischemic pain (Ischemic)
Where do get the answer to these questions?
This paper from Dr. Robert Formean(Ref 2) university of Oklahoma is just the best source I think, to explore and understand the topic. (Reading time 60 minutes: Let me tell you, it is worth more than a time spent on an insignificant angioplasty of painless PDA lesions)

Final message
So, what have you learned from this post? Does this question about angina matter at all? Surely not. in this space-age cardiac care where we are right inside the coronary even before we listen to the patient’s complaint properly. We are always at liberty to do what we want( or love) to do. But, the urge to understand the foundations of clinical science is the last remaining hope, that will keep the specialty of cardiology enchanting.













{kind=link}
{kind=link}