From kindergarten, we cherish stories. After adolescence, we dive into fiction. Then in college , science becomes sacred , and we believe, it is an eternal truth.
What a naive notion ? Science mirrors fiction and evolves relentlessly. Yesterday’s facts, become today’s fallacies with the stroke of a keyboard.
The myth of HDL as “good cholesterol” has been continuously propagated to the public and is etched in our (Patients & Physicians) minds. Likewise, LDL’s portrayal as the ultimate villain persists. Yet, emerging evidence flip-flops this narrative. Large, buoyant LDL particles appear harmless, even protective, while dysfunctional HDL may harbor hidden dangers.
This ignorance-based lipidology stems from oversimplified dogma, that ignores particle size, protien content, and its function. Large, fluffy LDL evades arterial infiltration, unlike small, dense variants. Meanwhile , dysfunctional HDL, oxidized or inflamed, loses its anti-atherogenic prowess and may promote oxidation and inflammation. (This may look like an exaggerated statement, but the fact that the largest popualtion at CAD risk : south asian metabolic syndrome, have a normal LDL level , tells us a chilling truth and mis- understanding about the lipid mediated CVD.
The curious paradox in dyslipdemia : It is the protien fraction that dictates the risk & benefit
In dyslipidemia, a key paradox exists. The protein fraction apolipoprotein that determine true risk and benefit, not lipid content alone.
ApoB-100
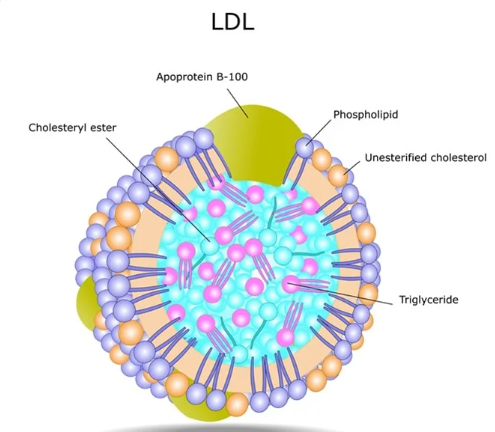
Apo B100 forms the structural backbone of atherogenic LDL. There is one Apo B 100 particles one molecule of LDL. It quantifies total harmful burden.Unlike LDL-C (which measures cholesterol load), ApoB-100 directly tallies particle count for superior CVD risk prediction. In large buoyant LDL, reduced ApoB-100 atherogenicity makes these particles largely benign and non-infiltrative.
ApoA-I/ApoA-II in HDL
ApoA-I (70% HDL protein) activates LCAT for cholesterol esterification and drives ABCA1-mediated efflux from macrophages.
It also suppresses LDL oxidation and vascular inflammation, embodying HDL’s core anti-atherogenic shield.
HDL structure
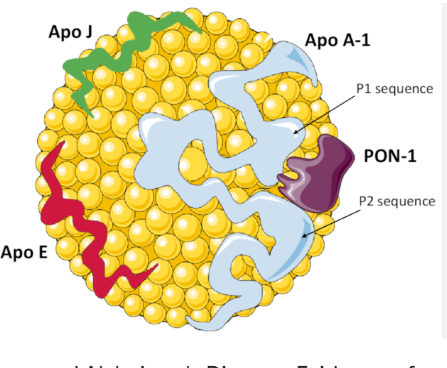
ApoA-II stabilizes HDL size/composition but impairs ApoA-I function in dysfunctional states converint HDLto a pro-inflammatory molecule. ApoB/ApoA-I ratio is key for the proper functioning of HDL. It is also proven, that the beneficial effect of HDL is lost beyond 60mg/dl.
Final message
Whenever we discuss hyperlipidemia, we falsely blame the lipids for endothelial injury .Realistically ,it is the protein sub-fragment Apo B 100 , which acts like a knife and hides within the lipid core , and attack the intact endothelium. There is no empty or toothless LDL molecule without Apo-B. However, there is a well known phenomenon of large bloated LDL , with excess foamy cholesterol,that sort of covers sharp edges of the Apo-B* like an umbrella and reduce the risk of endothelial injury.
*No reference : A pure logical Imagination.
Next query in queue : Are we sure , the statins,Rapathas & Inclisarans always reduce only the bad LDL ? (or it may reduce good LDL as well and give us a pseudo sense of bliss)
Find your own answer
Reference