No medical painting Impacted us this much. “The Doctor““was exhibited in 1891 by Sir Luke Fildes and Presented by Sir Henry Tate in 1894 in London. I am sure most of us are aware of this painting. Sharing it for future generations. Wish, this painting is embedded permanently in every young medico’s nimble brain.
This painting soulfully pours the silent and sacred bond between a helpless doctor and his dying patient, , which happens to be a lovely child.
Anyone who is flushed with millions can become a member of “The Hurlinghamin” London or the “Yacht” in Monaco, or any other glamorous clubs in the world. But, EBC* is different. Only cardiologists are privileged to enter. EBC is obviously unique. It is the only club addressing exclusively a subset CAD, ie bifurcation coronary lesions. Apart from immense pride and academic entertainment, it teaches us some deep technical points. (By the way, BFL* is a minuscule spoke in the gigantic wheels of global atherosclerosis)
Approach to BFL
90% of BFL still require only drugs or humble single stent or a provisional second stent strategy. However, as per basic rules of human intellect, lesser problems continue to bother us and consume our precise time. This continues, even after realisng , there are 6 complex two stent strategies that doubles up complications.
(*EBC -European Bifurcation club .BFL-Bifurcation lesion)
We will address one unique issue in BFL. May not be a major clinical issue .Still, it’s worth it. It is about the side branch crossing after the intentional jailing of the side branch.
Let us answer this query first
Pure science: Observations from Bench test (Text from Ref 1)
In contrast to provisional stenting in which re-crossing through a distal strut is desirable, initial re-crossing the crushed SB stent in the DK crush technique should be done through a proximal stent strut to minimise the risk of SB stent deformation. A theoretical exception could include bifurcations with a particularly narrow angle for which proximal re-crossing may shift more struts towards the MV, leading to a less desirable, longer neocarina. The 2nd recrossing should be done through a distal strut.
Bench testing is clear. Still , Why this confusion ?
We are all talking about theory in bench testing in stable non-hemodynamic conditions that lacks a biological carina and the dynamics of plaques. We are aware that there are at least 5 virtual ostia in every bifurcation arena (or trifurcation) . The qunatum and direction of plaque sharing occurs with a random effect. We also know the final kissing either cements these plaques perfectly or unsettles in a most bizarre way. So, these strut crossing stuff, are more of an imaginary bio-engineering principles. Whichever strut you cross, do it slowly, gently dilate it to the maximum within the polygonal confluence and good approximation. Don’t get too much carried away , even live online OCT guidance do not guarantee a perfect crossing.
Final message
The answer to the title question seems to be (me), one need not hair-split much on the site of crossing at the side branch. Fortunately, in whatever way, we weave the metallic mesh*, at the epicenter of the coronary highway, it is the natural secretagogues like TPa, Nitric oxide, PGI-2, along with DAPT decides the patient’s genomic fate.
*An appeal to all EBC club members.Before embarking upon a compex PCI on a stable patient , please think for one last time , whether your patient might do well, only with medicines.
Referecne
1.Hall AB, Chavez I, Garcia S, Gössl M, Poulose A, Sorajja P, Wang Y, Louvard Y, Chatzizisis YS, Banerjee S, Xenogiannis I, Burke MN, Brilakis ES; Collaborators. Double kissing crush bifurcation stenting: step-by-step troubleshooting. EuroIntervention. 2021 Jul 20;17(4):e317-e325. doi: 10.4244/EIJ-D-19-00721. PMID: 32310131; PMCID: PMC8919516.
AVR plus MVR commonly refered to as DVR is a path breaking cardiac surgery in patients with combined valvular heart disease. Still, it carries considerable mortabidity, if not done with high degree of expetise and standards . Apart from technical perfection, an inherent issue exist that might affect long term Impact.
Here is lateral view of X – ray chest showing a DVR.
Note :The green ring denotes the mandatory gap to avoid metal clash ( a missing biological link) in the Aorto mitral continuity
Final message
What will happen to the native anatomical and physiological functional unit of Aorto-mitral continuity after DVR? Will LV inflow encroach the LV outflow or vice versa? These questions are less often asked. In the process, we often ignore a concept called Aortic-mitral coupling or inter-dependence.
Stents have become essential coronary jewelry in global CAD care. The usage of these glamorous metals inside the human heart has crossed many millions, and aiming for a billion. Obviously, when used on such a large scale un-toward events are not an exception.
Stent migration
While the complication list of coronary stenting hangs long, fortunately, stent migration is a rare event. , Though, early geographical miss is common during deployment, these episodes don’t come under stent migration. Stent dislodgement during the procedure is a more common technical defect.If not recognized it may be wrongly labeled as late migration Exaggerated longitudinal shortening sometimes mimics migration.
Minor stent skids or mini migrations in cath labs are acceptable, distant and wayward migration to dangerous zones can be problematic.
True migration may be defined as a stent moving away from the target zone in the short or long term from its original location. It is useful and can be further divided with reference to time like peri procedural, short term, and late. Though it is generally thought to be rare true Incidence is not known. (Colkesen AY, . Int Heart J. 2007;48(2):129–136.)
Mechanism
It is not always easy to find the reason for migration. Many of the following factors could operate
It is tempting to blame the technique, inappropriate size (small for the lumen), and lack of imaging whenever migration occurs.(Very often one of them is true)
Metallurgically, it is the radial strength that keeps a stent in its original place. if for some reason this force is lost there is potential for migration of the stent. In this context, there is a link between stent fracture, injury infection, and migration of the stent.
A vasospastic coronary artery holding the stent tight and later releasing the grip is a rare possibility.
Liberal use of vasodilators like nitroglycerine might contribute. A case report from Pakistan suggests this possibility Murat Celik, Pak J Med Sci. 2013
The coronary artery is not a static tube so is the stent . the artery can milk out foreign bodies if it wishes. Fortunately, it doesn’t. Instead, it reacts with metal and initiates an inflammatory and rejection process that may ultimately end up as infective aneurysm and the stent can migrate outwards or extraluminal into the aneurysm sac. We realize, this is more common than other forms of migration.
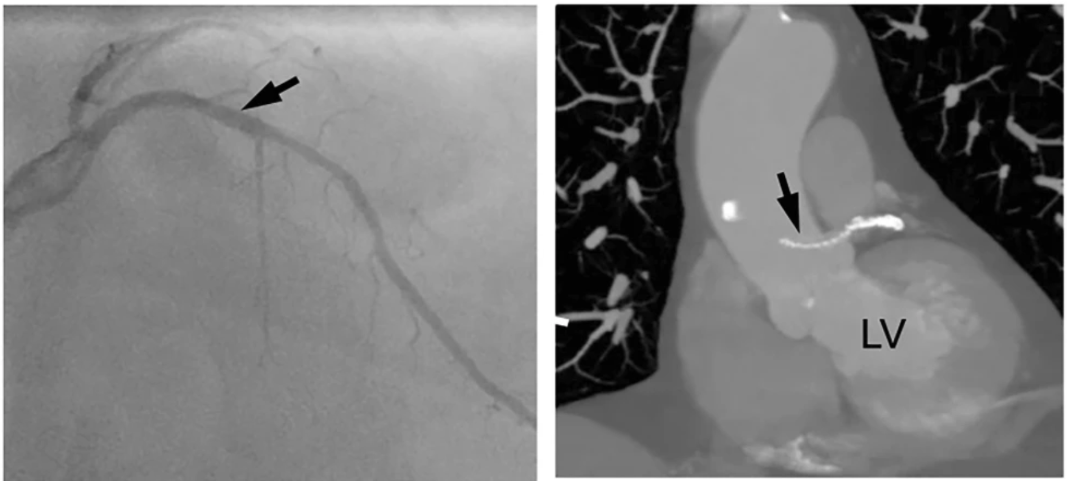
Retrograde migration is a baffling complication as in the following case.
A LAD stent migrates retrogradely into the aortic root crossing the left main ostium. Image source and courtesy Hilary Bews and others, Coronary stent on the move, European Heart Journal – Case Reports, Volume 5, Issue 12, December 2021, ytab511, https://doi.org/10.1093/ehjcr/ytab511
Implication
Stent migration can be totally unrecognized by many, while could end with a dramatic clinical event depending upon the extent and location of migration.
Though it is a sort of crisis for cardiologists, very often patients are asymptomatic and comfortable in spite of migration. (After all, it is the same pro-thrombotic foreign body even if it is present in its original place, is it not ?) Minor migration or embolization to safe zones need not require any intervention.
What should we do if leaving alone is not an option?
Crossing the stent and deploying it again or trapping or crushing it with another stent or retrieval are various options. Surgery is the last resort if the migrated stent is compromising blood flow critically.
Stent migration: A mini ethical crisis
Should we inform the patient about this adverse event?
Never hide any info from your patient about his or her health. It may amount to serious negligence. Sorry, I beg to differ*. If we really feel, it is a safe migration, and if the patient’s (& relatives) anxiety is too high, principles of practice of medicine can be selectively put on hold, for the overall benefit of the patient. (Of course, legal requirements are to be fulfilled by documenting the event in a complex manner as the lawyers do )
*Once you inform the patient, the option of leaving it unattended is a Herculean task even if it’s benign migration. (at least in our country) We have had long debates about this. Once upon a time, we had a stent that got dislodged and lost in circulation, and after long times of screening, there was a suggestion of a stent in the pelvic branches of the iliac artery. I will leave to your imagination, what we did for that patient.
Final message
Stents have conquered our profession and it is a life-sustaining device for both patients and cardiologists. Along with it, we have percutaneous valves, wires ,plugs, disc conduits, ICDs etc, Welcome to the big world of vascular foreign bodies. We are supposed to get optimally trained to keep all this stuff within the circulatory system smoothly running. If you look in that perspective, stent migration may be considered a minuscule untoward event. This doesn’t mean we can take this entity casually. We need to be familiar with its true incidence, mechanisms, potential solutions, and preventive strategies.
Reference
1.Bews and others, Coronary stent on the move, European Heart Journal – Case Reports, Volume 5, Issue 12, December 2021, ytab511, https://doi.org/10.1093/ehjcr/ytab511
2.Kasegaonkar AM, Chudgar P, Kamat N, Burkule N, Dhareshwar J, Dalal A. Delayed Presentation of Intracoronary Stent Migration in Pericardial Space: Role of Imaging. Indian Journal of Clinical Cardiology. 2022;3(4):209-210. doi:10.1177/26324636221087108
I think this child underwent successful surgical correction.
What is the mechanism of angina in coronary AV fistula ?
Coronary steal is easy explanation. But, very few of them develop functional Ischemia even at exertion. Why ? Size of fistula. dainage site, complex tortuous tracts, associated microvascular obstruction, contribute more.
Reference
Angelini P. Functionally significant versus intriguingly different coronary artery anatomy: anatomo-clinical correlations in coronary anomalies. G Ital Cardiol. 1999;29(6):607–615. [PubMed] [Google Scholar]
Cath lab nightmares may be a cliche word for all of us. Still, It has become a mandatory topic in any cardiology conferences and live workshops. These sessions are always popular and crowded as Interventional cardiologists are eager to get tips from other experts, on what to and what not to do in the cath lab in crisis situations. While stuck in an unexpected problem, these tips really help us come out of this, with shared expertise, presence of mind, skills, innovation, etc.
I asked them, what can be done about this ? “Nothing much” was the unanimous response
Final message
To end on a positive note, nothing is Impossible. Let us first start feeling this moral nightmare, and quell it at its origin. Fortunately, this doesn’t need sophisticated hardware. All we require is a little bit of righteous & peaceful application of mind in the way we learn and practice science.
Whenever possible ,before doing a coronary revascularisation procedure , check twice the segments you try to perfuse is really short of blood supply and truly needs the procedure. Don’t ever waste your resources and try to blood-feed the dead myocardium. It’ can never be awakened !
Pragmatic science
I was conversing with my colleague recently , who has grown into suave , Interventional cardiologist with a huge academic & societal repute .He owns a personal cathlab and planning to get one more.
I learnt a non-academic reality lesson from him .
When planning myocardial revasascularisation, apart from myocardial viablity status, there is one more viability issue which is done in the account books of finance mangers across big hospitals. Its Cath lab viablity. Trust me, he used exactly the same word ! He went on to explain in detail , how, every day there must be a minimum number of procedures to keep the machine alive. Which is under the eagle eyes of the guys who funded the state of the art lab !
“So, what do you say,I asked him ?”
He was frank enough to admit, he felt always happy when he is able to convert angiograms into angioplasties.He went on to add , the Ideal CAG-PCI conversion ration should be atleast 3:1 or more.
“Whenver I hear such genuine statments from real world people , it pains, as it tends to confirm my assumptions ”
Final message
I am wondering with all my lost wisdom. Why should any cardiologist after 30 years of training, fight for cath lab viablity , and get into a conflict with the very organ they are supposed to care and protect.
When did we become so Inferior beings & fight for the survival of these life less machines ?
Meanwhile, major text books , has un-intentionally facilitated this academic deciet .They have largely taken away the sting out of the snake . Myocardial viablity , hibernating, stunned myocardium , are rarely given importance nowadays and made it appear taboo concepts,in cardiology academia.
Postamble.
Will be extremely happy if what is portrayed in this post is not really true.
Having retired, find little more time in browsing the academic images lying idle in old computers.It is interesting, still a tiring job to pick any useful learning stuff, from heaps of data hiding in different hard drives.
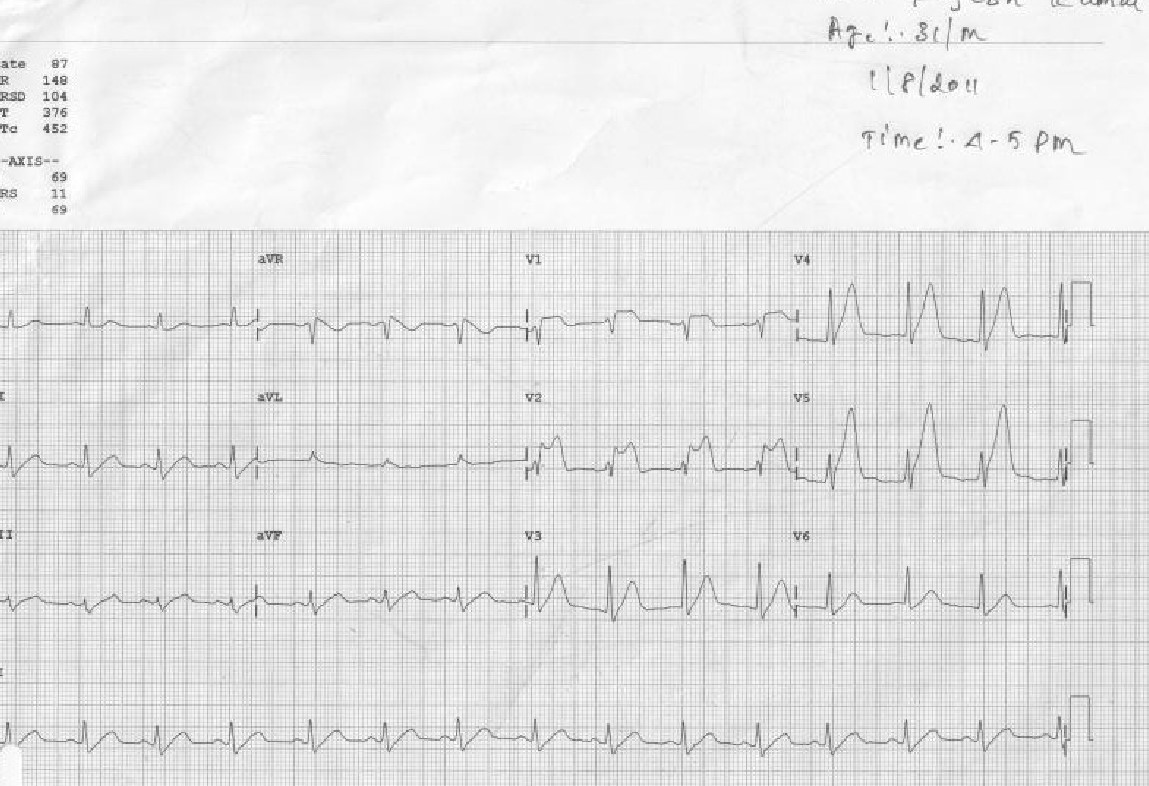
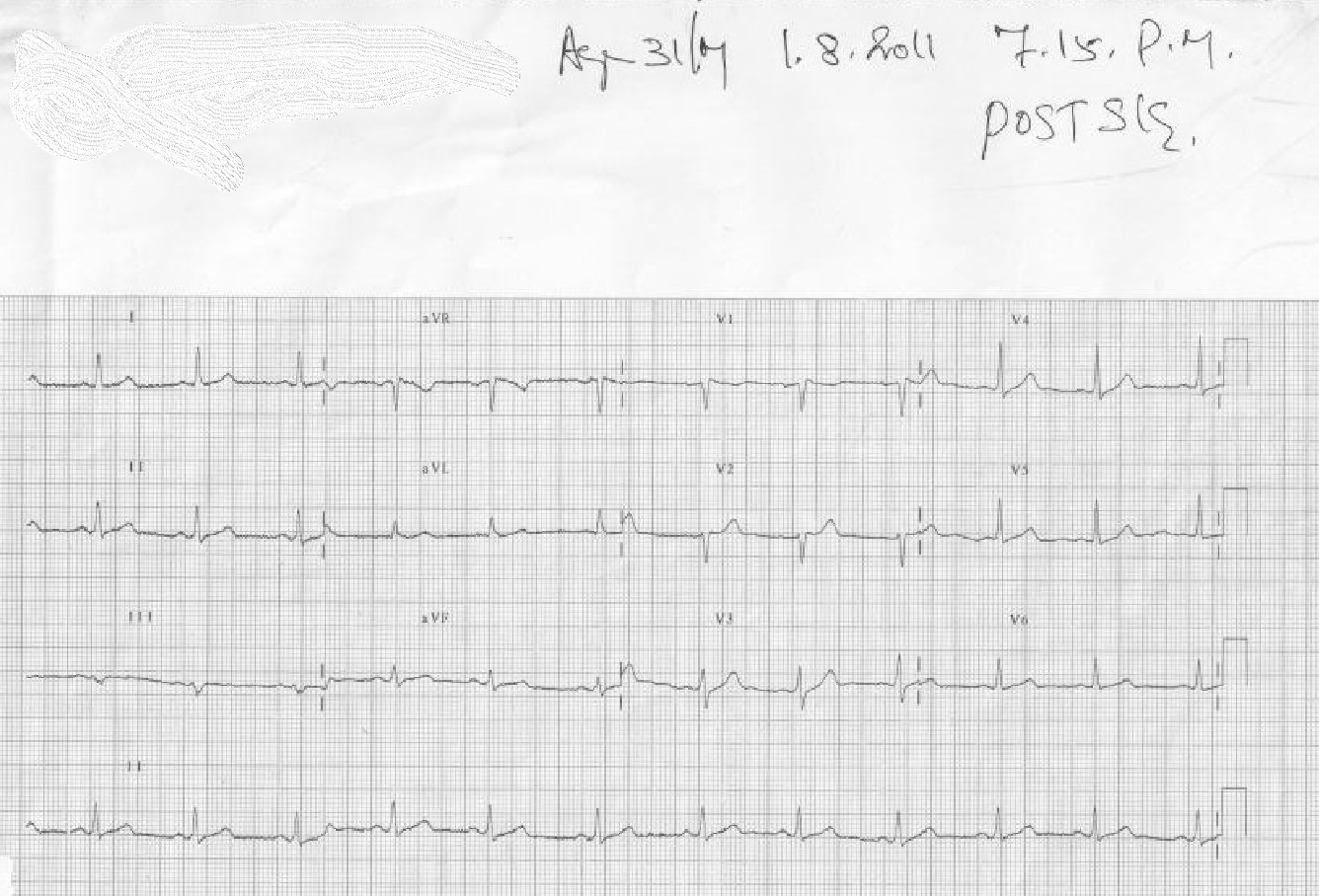
This set of ECGs I could retrive from the year 2011, A 31-year-old male presented to our CCU at 4.50 PM.
The treatment was Initiated in 10 minutes and completed in an hour, (Those days cath lab wasn’t functioning 24/7, more importantly, there was no external interference with our professional decision-making process)
The ECG was repeated at 7.15 PM
I think this case is much relevant even today, because it made me guilty of committing a crime*, by treating a STEMI without the help of a cath lab and discharging the patient with near normal ECG and LV function. The guilt was further amplified as I had used the lowly streptokinase, and not the glamorous Tenecteplase which could have produced a brisker and more complete TIMI 3 flow.
*One of my corporate friend called it a heinous one by current standards, for not attempting to visuvalise the IRA and a possible pharmaco invasive PCI.
Final message
STEMI can be tackled successfully in a number of different strategies. Immediate cath lab care is an optional accessory in the majority and of course, it can be life-saving in the minority. If we are unable to differentiate which patient will truly benefit from urgent cath lab intervention, I think, we have a huge problem, with the way we learn and teach cardiology. Hiding behind double-blinded statistics and RCTs is not going to bring a solution to this largely self-inflicted scientific predicament;
PH has always been an exciting academic topic in cardio-pulmonary medicine, for both clinicians and researchers. It is also one of the extensively studied hemodynamic parameter. The pressure in pulmonary circulation is intimately tied to the function of two critical organs. lungs , heart and various systemic factors. The fact that pulmonary circulation is essentially expansive & engulfed by the dynamics of lungs, makes PA pressure a continually variable parameter. Further ,the chest wall compliance, airway resistance , influence of pleural pressure fluctuations, make it difficult to estimate the normative pulmonary artery pressure and resistance even in rest.(Imagine during exercise !)
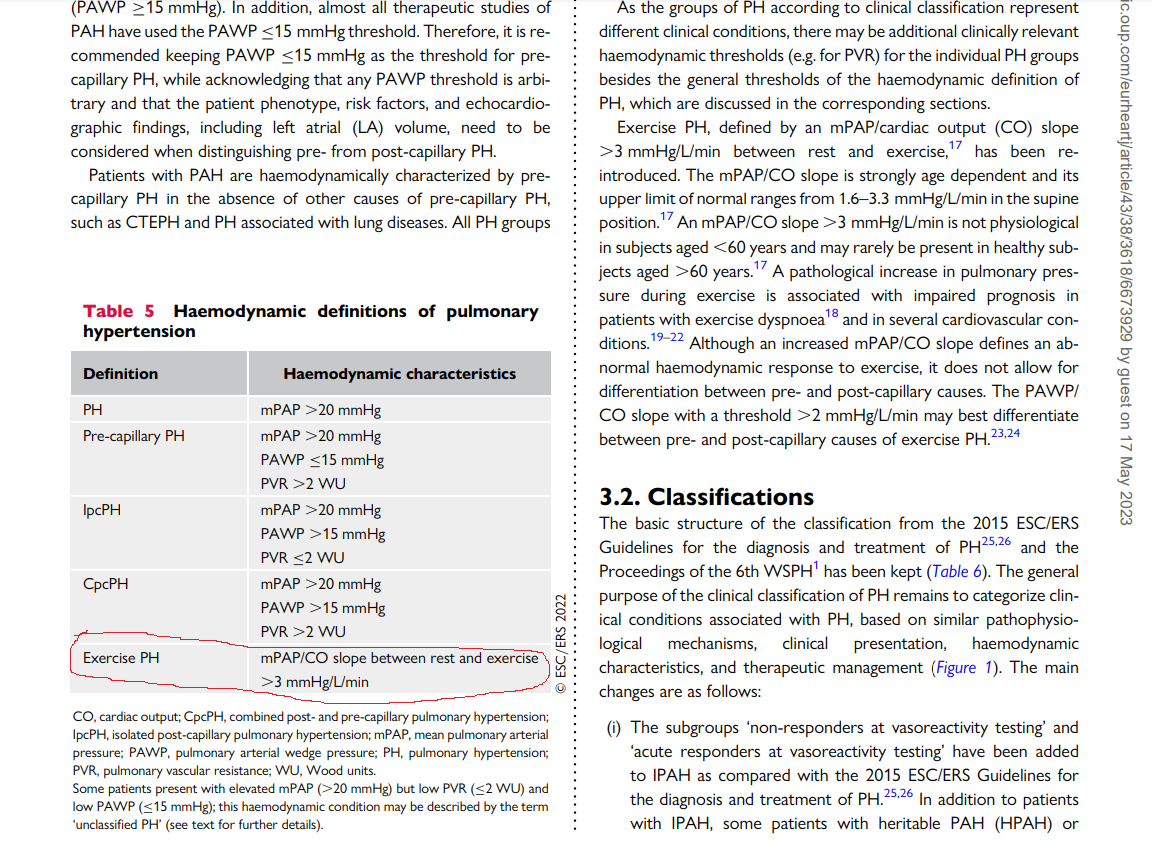
No surprise, our knowledge base about PH is under constant flux. The trouble starts with this query, What is the normal PA pressure ? After toying with various numbers we are currently hanging all our wisdom at a mean PA pressure > 20 mmhg as cut-off to define PH. However, we are able to grossly classify PH into various categories , pre/ post /combined etc. Here again, we have a guess work with two more cut offs.. For PCWP we have decided to choose 15mmhg over 12mmhg as upper limit of normal & PVR < 2 Wood units.
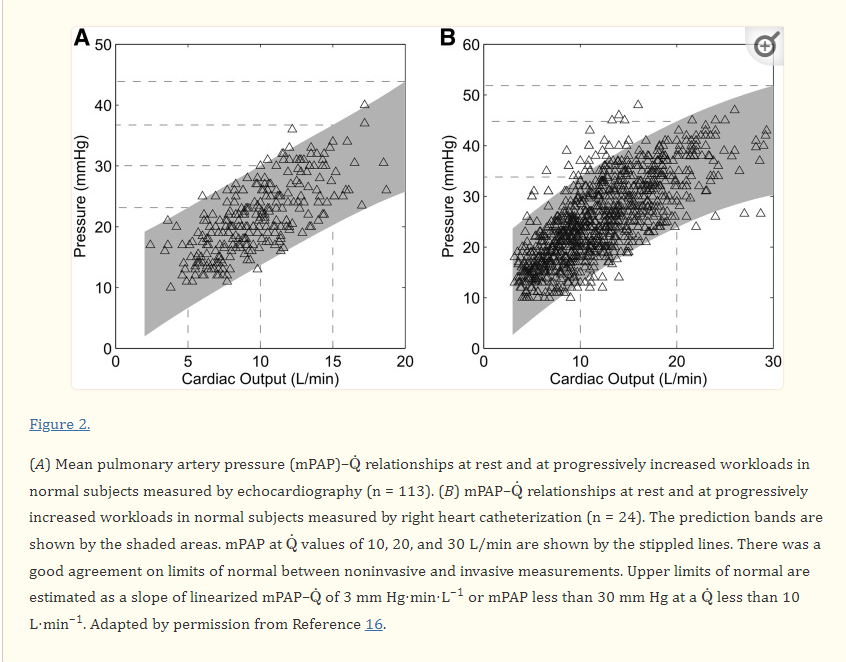
The second query in PH is still more contentious. What is the effect of exercise on PA pressure ? In our student days we were not allowed to bring exercise into the picture of pulmonary hypertension, in spite of the fact cardiac output increases up to 5 fold during peak exercise, Now, there is evidence to show exercise can increase PA pressure significantly, beyond the limits of current definition of PH. This is problematic for obvious reasons. Still, there has been considerable reluctance to accept exercise induced PH as a clinical problem by many of us .
*To be fair with our intellect, I think, we haven’t yet approved “Exercise induced systemic HT” as an entity officially. (Of course, hypertensive response during stress test is well known)
Seeds of New thinking
Thanks to current guidelines from ESC in 2022 .The exercise induced PH has come back with a bang and finds a place right behind the pre and post capillary PH. (See below ) I am sure, there must have been a vigorous debate before including this in the definition. We must appreciate the authors of two forgotten papers for the major shift in our understanding .(Ref 1 ,2)
The secret of the slope : From where did it come ?
ePH is > 3mmhg /Litter/Minute is the definition of ePH
It is the rate of raise that matters not the absolute pressure. This slope was validified in by Bossone E et al (Ref 2)
Some questions on ePH
1.How do you define ePH ?
Mind you, it is not an absolute number. It is the slope more than 3mmhg per litre of cardiac output. I agree to measure the slope > 3mmhg we need serial measurement and may be impractical .(Immediate post exercise echo is a close alternate )
2.Why we depend on slope rather than absolute value ?
This is because during heavy exercise PA pressure can raise even up to 30 or 40 crossing the boundaries of PH ..Only the rate of raise ie the slope can tell us whether it is appropriate or inappropriate.
3.Does ePH is really a clinical problem ?
Yes. it should be suspected in every unexplained dyspnea .(Beware of the anxiety it may elicit to the patient, so, go slow with your investigation first rule is to rule out Anemia and other common causes )
4.Can ePH occur over and above established causes of PH ?
Why not ? it is very well possible.(PH before and after six minute walk test will unmask this component)
5.Can we further classify ePH ? (Pre vs Post cap ePH)
Possible yes. ePH can be a marker of HFpEF if LVEDP is also correspondingly increased or else it will fall in to CETP or COPD.
6.Can COPD cause ePH ?
Yes, possible.
7.How does RV function confound ePH ?
This is ticky. Perfect RV-PA coupling and a good RV function is required to sustain ePH. A poorly contracting RV will make the whole concept of ePH and the defining criteria redundant. May be, we need to work for RV function corrected ePH . (This is a potential research topic for fellows)
8.Where do diastolic stress testing fit in diagnosing ePH ?
In one aspect ,DST which is screening test for silent HFpEF is an example for subtype of ePH.
Final message
The concept of ePH has entered once again into the cardio pulmonary clinical domain. Thanks to ESC 2002 team for listing this hitherto ignored disorder. Let us reiterate the importance of this concept in the clinical practice. It is worth considering some form of stress test to recognise this entity, in every patient who has unexplained dyspnoea.
The contents of the this blog is being published as Kindle E book , as per the request of many of the readers. Every article will continue to be open source in this site. Again I shall reiterate the book format is not aimed at any commercial intent. It is only to facilitate learning in a single book format Here is the link to book https://amzn.in/d/euhL5vu
Click below to see who is watching this website live !
This site will never aim for profit. Still ,this donation link is added at the request of few visitors who wanted to contribute and of-course that will help make it sustainable .
Please Note
The author acknowledges all the queries posted by the readers and wishes to answer them .Due to logistic reasons only few could be responded. Inconvenience caused is regretted.