It was April 15th 1912, Titanic, the Invincible, had just sunk into the dark waters of the Atlantic coast off Newfoundland. Exactly same time around, Dr. James Herrick, In Chicago, Illinois was busy documenting the first diagnosed case of acute coronary thrombosis. A new disease was born ie Myocardial Infarction. This was also the era of the Noble Prize-winning Invention of the ECG machine by Waller, Einthoven, and Thomas Lewis & co that sow the seeds for the specialty of electro-cardiology.
Though much was studied about MI with pathological specimens in the subsequent decades, there was a lull in the efforts to define the entity of myocardial Infarction till WHO defined in the early 1970s. It was dogmatic, still fair enough. (Clinical, Enzymes, ECG criteria, with any two feature, must be present to diagnose )
Since then, the field of cardiology has seen unprecedented development in both the diagnosis and treatment of ACS. We now have a universal definition( EHJ 2019 Thygesen K ) that asks us to triage based on high sensitive troponin followed by clinical and other parameters. STEMI usually doesn’t have much diagnostic confusion.
Nomenclature Issues in NSTEMI/UA
The definition of NSTEMI refuses to settle, though we have come a long way since the times UA/NSTEMI were clubbed together as siblings. The term unstable angina was coined by one of the most revered cardiologists of our times Dr. Noble O Fowler in 1978. They are the same one hitherto referred to as Intermediate coronary syndrome/Pre Infarction angina. Later, if enzymes were raised it was labeled as non-transmural/Non-Q MI. This became the classical NSTEMI later changed to NSTEACS (Still it is valid)
The semantics surrounding the NSTEMI is unlikely to end as long as we depend largely on ECG to diagnose and treat complex coronary obstructive syndromes. This, by no means, undermine the importance of ECG in this setting. It will remain the gold standard as far as, I can look into the future.
Some observation about the new ESC 2020 NSTEMI guidelines
Anyway, ESC 2020 has addressed this issue. It suggests a new term “ACS without persistent ST elevation” for NSTEMI (Ideally they should have used this abbreviation NP-STEACS)
(*I guess, the current ESC 2020 guidelines really wanted to get rid of both NSTEMI/NSTEACS for a very valid reason but still it was worried about the confusion it might create so retained the old term NSTEMI/NSTEACS )
The categories included in the current NSTEMI scheme are
1.Transient ST elevation (How transient ? Prinzmetal/ Non Prinzmetal ?)
2.Persistent ST depression
3.T inversion
4.Flat (Absent ) T wave
5.Pseudo normalization of T
It may include the following as well (Not in official ESC 20220 guidelines)
6*.Hyperacute T (Very early STEMI ? or NSTEMI?
7*.Wellen/Dewinter or its variants
I think ESC is to be appreciated for recognizing an off ignored observation that UA may have a transient ST elevation and end up later as NSTEMI/NSTACS. This group of ACS still poses a challenge for us to understand the overlap between total and subtotal coronary occlusion (Non-Prinzmetal ST elevation)
Final message
Does this nomenclature issue create problems in management?
- Yes, it does. The major implication is in the diagnosis ACS with dynamic ST segments ( ST-elevation / /depression or any combination)
- If a probable STEMI after spontaneous lysis presents as NSTEMI, Is it the baby STEMI or neo NSTEMI ? One may not rush such NSTEMI patients to cath labs.
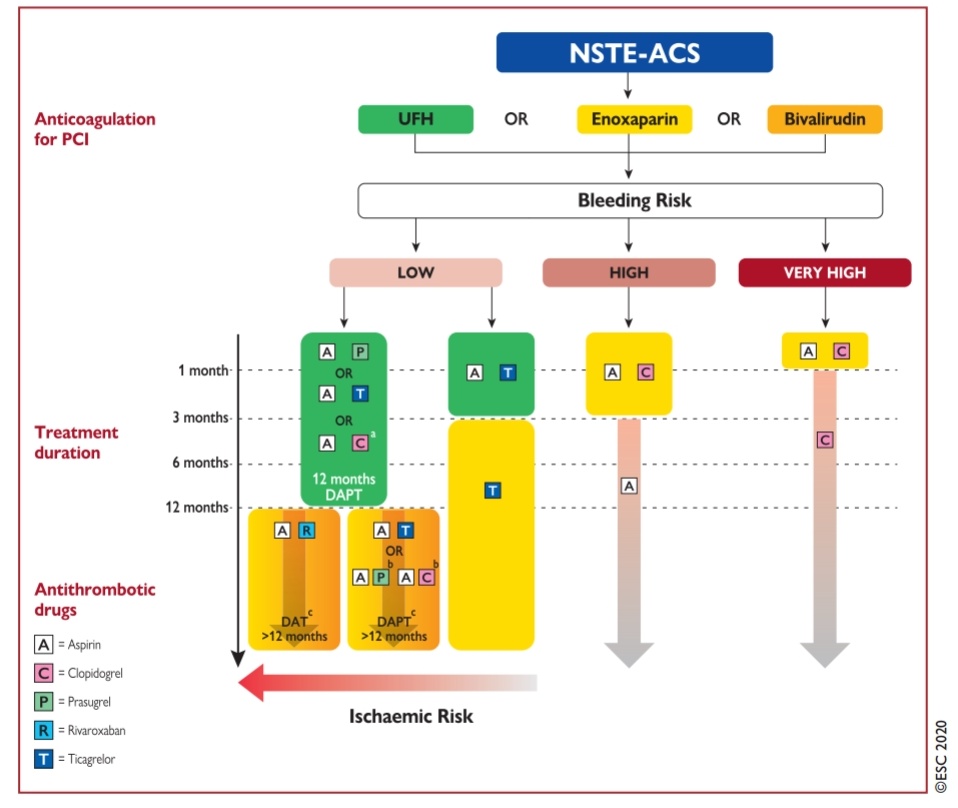
- Of course, many of us are conditioned to follow a “single point agenda “ that dictates all ACS shall reach the cath lab and managed thereafter based on coronary anatomy. If that is the case, I am sure the bulk of this 79-page new NSTEMI guideline appears redundant.(Ref 1)
Reference
1.Jean-Philippe Collet, ESC Scientific Document Group, 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The Task Force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC), European Heart Journal, , ehaa575, https://doi.org/10.1093/eurheartj/ehaa575
2 Fourth Universal Definition of Myocardial Infarction (2018). Eur Heart J 2019;40:237-269.