There are many cardiology journals we read , trust , and celebrate . . .
Many of us are not aware of few other excellent journals
This is one is different
It is from Scandinavia & deserves a special status.

Posted in Uncategorized, tagged cardiology, cardiology journals, Scandinavian journal on February 21, 2010| Leave a Comment »
There are many cardiology journals we read , trust , and celebrate . . .
Many of us are not aware of few other excellent journals
This is one is different
It is from Scandinavia & deserves a special status.
Posted in echocardiography, Great websites in cardiology, tagged best cardiology website, doppler echocardiography, echocardiography, tee, transesophageal echo on February 21, 2010| Leave a Comment »
Internet is a wonderful gift for for mankind but only occasionally we find great resources .
Hats off to Dr .Pybus from Australia for his efforts
A must read for all cardiologists rather everyone involved with echocardiography
Click on the Image to reach the site
![]()
Posted in Cardiology - Clinical, Cardiology -Interventional -PCI, cardiology -Therapeutics, Cardiology -unresolved questions, cardiology- coronary care, tagged alteplase, pharmacokinetics, stemi, streptokinase, streptokinase vs tenektepalce, streptokinase vs tnktpa, tenektepalce vs streptokinase, tenekteplace, thrombolysis, thrombolytic therapy, tnk tpa, tpa vs streptokinase on February 20, 2010| 1 Comment »
Thrombolytic therapy was a mini revolution when it was introduced two decades ago .It has since evolved , not only in the molecular structure but also in it’s usage pattern.
The first generation streptokinase is continued to be used even today . While the latest generation thrombolytic agent TNKTPA(Tenekteplase) is threatening to push the old warrior out of CCU.

(Of course the American Physician & Pharma community never gave the due respect to streptokinase !)
The two common indications for thrombolytic therapy are
Uncommon indications
From the beginning , there has been a controversy about the thrombolytic dosage and the speed with which it is to be administered .Let us recall , streptokinase was initially used in various regimes ( 5-30lakh units between a 10 -3hr infusion ) Later ,we arrived at a consensus at 15L units in 1 hr infusion . TPA also experienced the same . Which settled for front loaded regimen(35 + 65mg) . The confusion reappeared when we developed bolus thrombolytic agents( TNKTPA) .
In STEMI thrombus formation is often a one time process while thrombolysis is a continuous process. In pulmonary embolism both thrombus formation and lysis is often continuous process .
The success of thrombolysis depends on the sustained drug concentration , the pressure at which the drug interacts the thrombus.
Many times it is prudent to administer intensive heparin after thrombolysis to prevent recurrent thrombosis. Further , most of the pulmonary embolisms will require long term anticoagulants.
How to maximize the success of thrombolytic agents ?
It makes sense , to administer these thrombolytic agents over a prolonged period of time so that the lytic process gets wider recruitment of the natural lytic mechanisms.

When a drug is infused continuously , the drug reach the thrombus in a pulsatile manner , which facilitates thrombus dessication (Like drip irrigation ) . A long acting drug even with a high concentration may not be very effective , since the drug is required to produce a mechanical effect here . (Unlike say a long acting antibiotics !)
TPA in Pulmonary embolism
The inadequacies of 2 hour infusion of TPA is glaring in acute pulmonary embolism .We believe a 48-72 hour streptokinase infusion has a definte edge over a short and brief TPA infusion.
Issues need answer
It is yet , not understood why we can’ t infuse TPA as a long term infusion like streptokinase .
Advantage of bolus TNK TPA in pre-hospital phase of STEMI
The argument in favor of bolus dose thrombolytic agent is the ease of administration .
The other the major advantage claimed is , a 10 second TNK TPA in STEMI can substantially reduce the time window and facilitate early completion of thrombolysis .
Counter point
But , the later concept is hard to prove . . .
In fact , there are no controlled studies available for assessing the efficacy of TNK-TPA vs Streptokinase with reference to various time windows. We presume so many things. An incomplete early thrombolysis may not be better than a more successful but slightly delayed TIMI3 flow .
As scientists, when we try to answer these question we ask for data . Are we getting it any way ? Are the existing data reflect fact ? We wonder, will we may never get an hourly angiographic data base about the IRA patency in TPA bolus vs streptokinase infusion .
It is most unfortunate, with many of the critical questions still to be answered , the cardiology community believes , they have reached the summit of knowledge about thrombolytic therapy . Current perception is , the research on existing thrombolytic drugs is deemed to have been complete .
In this hyped era of interventional coronary care , it is a remote possibility to have any further comparative studies on thrombolytic agents .
The greatest threat faced by us today is the destiny of modern medicine is often decided in few corporate board rooms and hence research questions rarely emanate from bed side !
In this scenario, where we are not likely to generate genuine clinical data , the only way to move forward is to go by our experience – ” Genuine experience to be precise . . .”
Final message
Ease of administration should never be the criteria in choosing a thrombolytic agent . It can severely compromise the quality of thrombolysis ! especially in pulmonary embolism and to a certain extent in STEMI. Success rarely comes with ease . . .

Many believe , the choice between streptokinase & TPA goes much beyond it’s academic reasons. TNK TPA (Tenektepalse) has come in a big way to replace streptokinase even in developing countries. Ofcourse it is backed by a huge study ! (ASSENT) .
The cost effectiveness and worthiness of TPA over streptokinase was never proved comprehensively.
Note of caution :
The observation made above is based on personal opinion in about 20 patients . Readers are argued to do their own analysis on this issue and come to a conclusion .
Posted in Cardiology - Clinical, cardiology congenital heart disese, tagged arcapa on February 17, 2010| 2 Comments »
Coronary artery anomalies are relatively common . It can be either in it’s origin, course , or termination etc.
There are two major sub groups.
The second category which we encounter in cath labs frequently does not have major implications . RCA and LCA arising away from it’s respective sinuses ,Separate origin for LCX, or conus, RCA from left sinus or a high take off of RCA are the common anomalies.
While coronary anomalies are commonly associated in complex congenital heart disease (TOF, DORV, TGV, etc )
Isolated complex anomalies of coronary arteries are extremely rare
This happens , when one coronary artery arises from pulmonary artery instead of aorta and it becomes a fascinating disease !
The ALCAPAs and ARCAPAs
When the LCA originates from PA it becomes a rare cause of left to right shunt .it is referred to anomalous origin of LCA from PA (ALCAPA) .
The ALCAPA is many times common than the “ARCAPA”
We report a case of ARCAPA (Anomalous orgin of RCA from PA )
The unique features of ARCAPA could be
Image and video of the ARCAPA will be uploaded shortly
Reference
1 http://asianannals.ctsnetjournals.org
2 http://ats.ctsnetjournals.org
Posted in cardiology -ECG, Cardiology -Interventional -PCI, cardiology -Therapeutics, Cardiology -unresolved questions, Uncategorized, tagged ECG on February 15, 2010| Leave a Comment »
It is said every clinical diagnosis needs to be substantiated with documented objective evidence .
Probably, the commonest cardiac emergency , that can be diagnosed purely by history is UA.
Yes , unstable angina is a symptom not a disease entity !
By definition UA is
If you read the definition again, you will realise ECG or enzymes never come into the diagnostic picture .UA can be diagnosed even before one has a look at the ECG ! So, it is too obvious one can diagnose UA irrespective of whatever is recorded in the ECG. Normal ECG is one such possibility.
When a patient is having severe compromise in the blood supply to his / her heart , how on earth , it is possible to have a normal ECG ?
It only tells us, ECG is not a fool proof method to exclude ongoing ischemia . When we know , ECG can miss even a STEMI it is not a big deal it misses a UA.
Apart from the electrical blind spots of conventional 12 lead ECG, following are the other explanations offered for a normal ECG in UA.We know UA occurs with ST depression(Classical ) , T inversion, rarely ST eelvation
So UA can occur with
Final message
Even though UA CAN occur with normal ECG , we are uncomfortable to diagnose UA without documenting ECG changes . We should realise this fact , as missing a diagnosis of UA , just beause the ECG is normal could have very costly consequence !
Posted in Cardiology - Clinical, Cardiology - Electrophysiology -Pacemaker, cardiology -ECG, Cardiology -Interventional -PCI, cardiology -Therapeutics, Cardiology-Arrhythmias, Cardiology-Coronary artery disese, Infrequently asked questions in cardiology (iFAQs), tagged cardiac action potential, ECG, stemi on February 14, 2010| 1 Comment »
STEMI is the commonest cardiac emergency . Many believe , we are close to conquering it . It is hardly the truth .
Here is a case history and ECG of a patient with STEMI .
 After thrombolysis , the paradox happened . ST elevation increased by 4mm and soon the patient became restless with worsening pain and became silent instantaneously , with monitor showing EMD and asystole .A diagnosis of free wall rupture was made.
After thrombolysis , the paradox happened . ST elevation increased by 4mm and soon the patient became restless with worsening pain and became silent instantaneously , with monitor showing EMD and asystole .A diagnosis of free wall rupture was made.

What we used refer in our CCU (Madras medical college Chennai .One of the oldest CCU in South Asia )
as “Action pontentialisation” of surface ECG . This ECG finding has great clinical significance .
Here is a zoomed up view of a qrs complex of the patient , which is very
closely resembles an action potential

 Picture courtesey http://ocw.tufts.edu/Content/50/lecturenotes/634488/634591
Picture courtesey http://ocw.tufts.edu/Content/50/lecturenotes/634488/634591
Pathological basis of “Action potenial” Like ECG
This heavy downpour of electrical energy that emanate from the myocardium means two things
Clinical correlates of action potential ECG
The death happens by a sudden rupture , EMD and asystole .
Can a life be saved by the much fancied Emergency PCI ?
Not really. The PCI can not reverse the myocardial damage , so it’s role is little . But , any way it should be done and . . . it will be done in most institutions to give the benefit of doubt (Of course , with a definite the risk of doubting !)
What is the risk of PCI in these situation ?
The infarct related artery * if opened up can convert a bland infarct into a “angry red” hemorrhagic infarct .This is as good as giving the patient , a farewell party for his journey to heaven !
Note : Primary PCI definitely saves life in STMI . The * is applicable only in persistent ST elevation , late after an acute MI.
How could have the above death prevented ?
As one of the comments to this article suggested, we need to have methods to identify impending rupture early and accurately .This should followed by a prophylactic surgical intervention (Reinforcing the friable myocardium – with a patch or mesh ) .This is again not a easy decision to make .
Final message
When the ECG assumes a shape of an action potential , it is often a sign of imminent death . Even though it may sound a pessimistic view it is often the truth . Of course , an emrgency PCI or CABG are the only options available , we have to be remember the above truth , as we play those sophisticated games within their coronary arteries.
Posted in Cardiology - Clinical, cardiology -ECG, Tutorial in clinical cardiology, tagged av block, bi fascicualr block, bundle of his, fascicular blocks, hemiblocks, lafb, lafb vs lpfb, left anterior fascicular block, lpfb, rosenbaums block, why lafb is more common on February 11, 2010| Leave a Comment »
The bundle of his divides into two
Middle fascicle * Many dispute it’s presence . One may wonder , how can anatomy be under dispute ? If you cut a heart you should be able to clear the dispute . But medicine is not that simple . . . What you do not see may be more important than what we see.
The anterior fascicular block (LAFB) is one of the common conduction disorder. It ‘s significance : Can be a benign or a dangerous entity depending upon the clinical situation .The problem with LAFB is , it is diagnosed primarily by the axis shift it inflicts on the QRS complex.
In a strict sense, it is not a ideal way .There is a tendency to label all significant left axis (> -60*) deviations as LAFB. This practice has made diagnosing LAFB very common in elderly, hypertensives, etc. In these situations it may not mean anything , except to suggest a delay in conduction in left anterior fascicle.
If we filter out all these benign axis shift ECGs , the true organic pathological LAFB may not be that common .
Organic , LAFB occurs in the following situations.
Even in degenerative , ischemic conduction defects LAFB is far more common than LPFB why ?
The traditional explanations are
** Which experiences the peak LV pressure at > 100mhg and a dp/dt up to 2000mmhg (While, the posterior fasicle is located away in the inflow portion of LV , which is exposed to low pressure – at best 10mmhg filling pressure )
Posted in Cardiology - Clinical, cardiology -ECG, cardiology -Therapeutics, Cardiology -unresolved questions, tagged acs, acute coronary syndrome, cardiologists, cva, deep t wave inversion, ECG, intra cerebral hemorrhage, mimicers of stemi, neurologist, non ischemic st depression, non specific st segemnt, qt interval, stroke, sub arachnoid hemorrhage, t inversion, u wave on February 11, 2010| Leave a Comment »
Human body is now approached by many of the physicians as collection of multiple organs . This is the price we pay for modernity in medical science. The era of great physicians in general medicine has gone . Now, a super specialist of one organ is rarely concerned about what is happening to the patient’s other organ , it is considered foreign to him ! While , this is the dominant thinking pattern of modern-day specialist
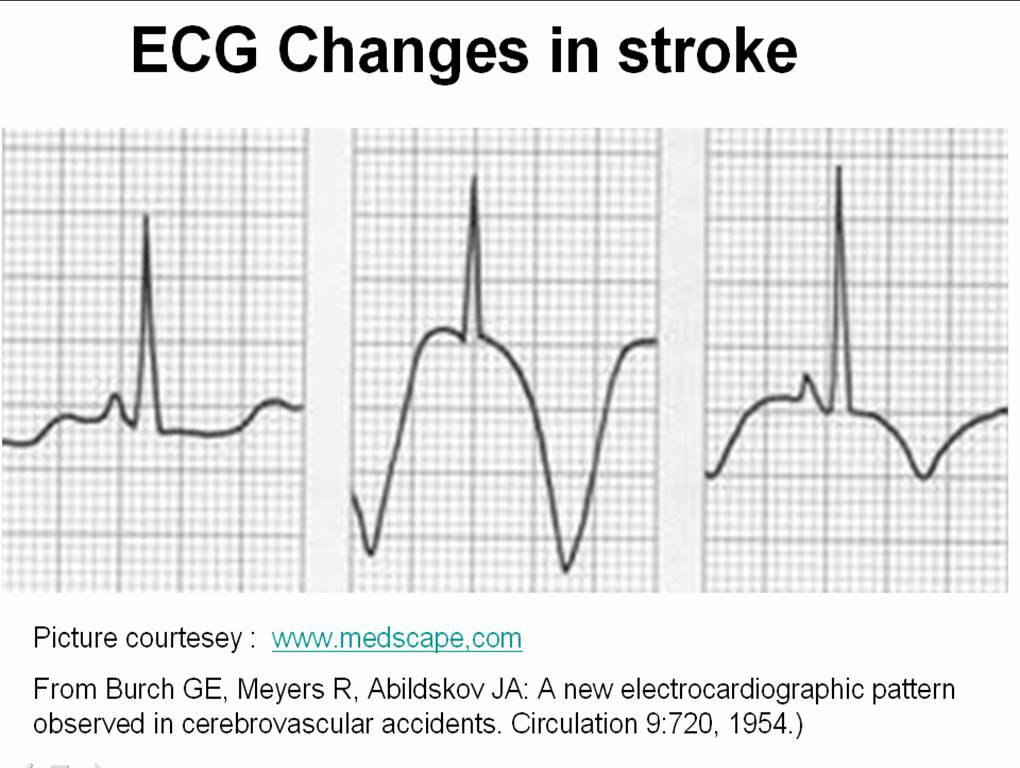
Let us travel intime and go to the year 1954 . . .
Three physicians from Michigan ,USA published one of greatest observation in clinical sciences , namely the ECG changes in various forms of stroke .
Now , a shrewd physician , will suspect a subarachnoid hemorrhage (SAH) by looking at the ECG when the clinical situation demands . But , what we need is every one should develop that skill . We have seen errors happening even in big institutions (or is it because it is big ?) when an elderly person comes with deep T inversions with or without altered sensorium being rushed into CCUs & cath labs instead of neurology units.
We need to teach our junior colleagues . . . That , ECGs of patients with acute neurological syndromes (ANS) can mimic as acute coronary syndromes (ACS) ( especially in elderly ) .
The following ECG changes * are observed during stroke

The ECG changes tend to occur very early after CNS injury.May last up to 1 week. They are not useful to identify the type of stroke. But , deep T wave inversions strongly suggest SAH rather than ICH or thrombotic stroke.
What is the mechanism of these ECG changes ?
It is a clear proof that heart and brain are interconnected by neural network. All the noted changes occur during myocardial repolarisation . (ie ST segment ) The current thinking is (Ofcourse , it is same as our thinking in 1950s !) it is mediated by adreneergic surge initiated by CNS insult transmitted to myocardium by the sympathetic system.
Why should SAH produce more ECG changes than others ?
It is possible the net adrenegic drive from the brainstem and spinal cord will be greater in SAH as it spreads the entire CNS through the cerbro spinal fluid. While localised ICH and infarct is likely to generate less adrenergic impulse.
Reference
Read the link to circulation 1964 .With courtesey to circualtionaha.com
http://circ.ahajournals.org/cgi/reprint/9/5/719.pdf
This came 50 years ago , we still quote their work and no one has improved their work .
Final message
If only we make the clinical bed side teaching as a regualr habit , we do justice to our great physicians of the past , who enriched our life with their clinical skills and passion for knowledge sharing .
Posted in cardiology -Therapeutics, Cardiology -unresolved questions, Cardiology-Coronary artery disese, echocardiography, Infrequently asked questions in cardiology (iFAQs), Uncategorized, tagged cardiac failure, cardiology, DCM, dilated cardiomyopathy on February 9, 2010| Leave a Comment »
As the name suggests dilated cardiomyopathy would imply cardiac chambers will dilate , at least some time in the course of the disease .It can be minimal, mild or massive. A new entity called non dilated cardiomyopathy is also gaining wider acceptance . (That will be dealt seperately )
Logic would suggest , the first chamber to dilate in DCM should be the left ventricle because it is facing the direct load of systemic blood. But we also know , whenever LV is stressed , left atrium comes to it’s assistance .
Left atrium does this by total self sacrifice ( by all means!) increases it’s force of contraction, elevating it’s mean pressure or even increasing it’s rate (AF) .
Like most other critical questions in cardiology , the factors that determine LV dilatation in DCM , is also poorly understood !
When the issue is complex , it is usual to make the the unknown genetic defects , the scapegoat !
As of now the most important determinant of LV dilatation could be the behavior of the desmins, the gap junctions and myosins the titins etc
If the LV of a DCM patient refuses or resists dilatation what might happen ? Is it good or bad for the patient ?
Here is a catch . A LV that does not dilate obviously should be be good for the patient is in’t ? Medicine is not that simple.
When LV fails to dilate it means it has become too stiff and rigid and pass on the burden to to LA which faces the music. And in the process it dilates.This is the reason , we observe diastolic dysfunction in vast number of DCM patients.( Currently it is estimated > 75% DCM will have significant diastolic dysfunction )
So , now we can imagine how complex the sequence of hemodynamic stress in DCM that determine the chamber enlargement.( RA, RV dimension in DCM is a separate issue !)
So now answer this question : Which chamber dilates first in DCM ?
The answer must be 3 .
Why recognising this sequence of chamber enlargement in DCM is important ?
Posted in Cardiology - Clinical, cardiology -Therapeutics, echocardiography, Tutorial in clinical cardiology, tagged aortic cusps, coroanry ostium, coroanry sinus, coronary, coronary artery, coronary artery origin, echocardiography, lcc, left main coroanry artery, left main disease, lmd, ncc, non coronary cusp, rcc, right coroanry cusp, short axis in echocardiography on February 9, 2010| 1 Comment »
Imaging coroanry artery is generally in the domain of interventional cardiologists. MDCT has helped us to change that.
The humble echocardiography can identify the origin* of coronary arteries in most persons. The resolution power of modern day echocardiography is 2mm and the left main ostium is >3.5mm in 99% of population . If some body says one can’t visualise the coronary artery by echo , it can only reflect their ignorance or lack of patience to get an optimal image. Of course technological limitations are there.
* To be emphasised again , only the origin can be identified.
Can we identify ostial leftmain or proximal left main disease by echocardiography ?
It should be possible in few .

Can we place a doppler sample volume within the left main and measure coronary flow velocity ?
When obsterticians are able to assess the uterine artery flow in a bulky uterus , it should be possible to do the same in a coronary artery . Motion artifacts is the issue in the heart. Micro sample voulme (<1mm) are expected in the future that will make a non invasive coronary flow assesment a distinct possibility.




{kind=link}