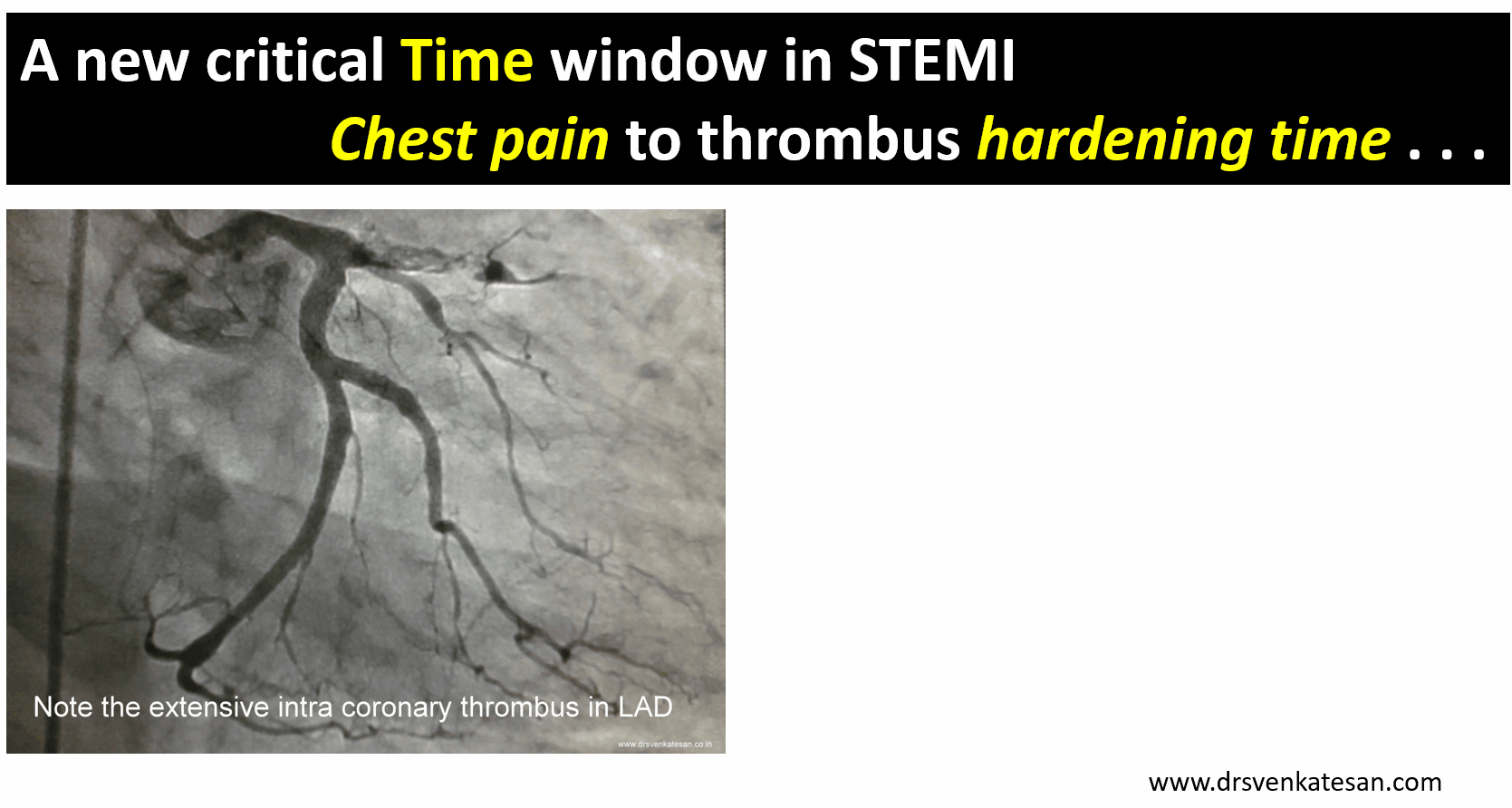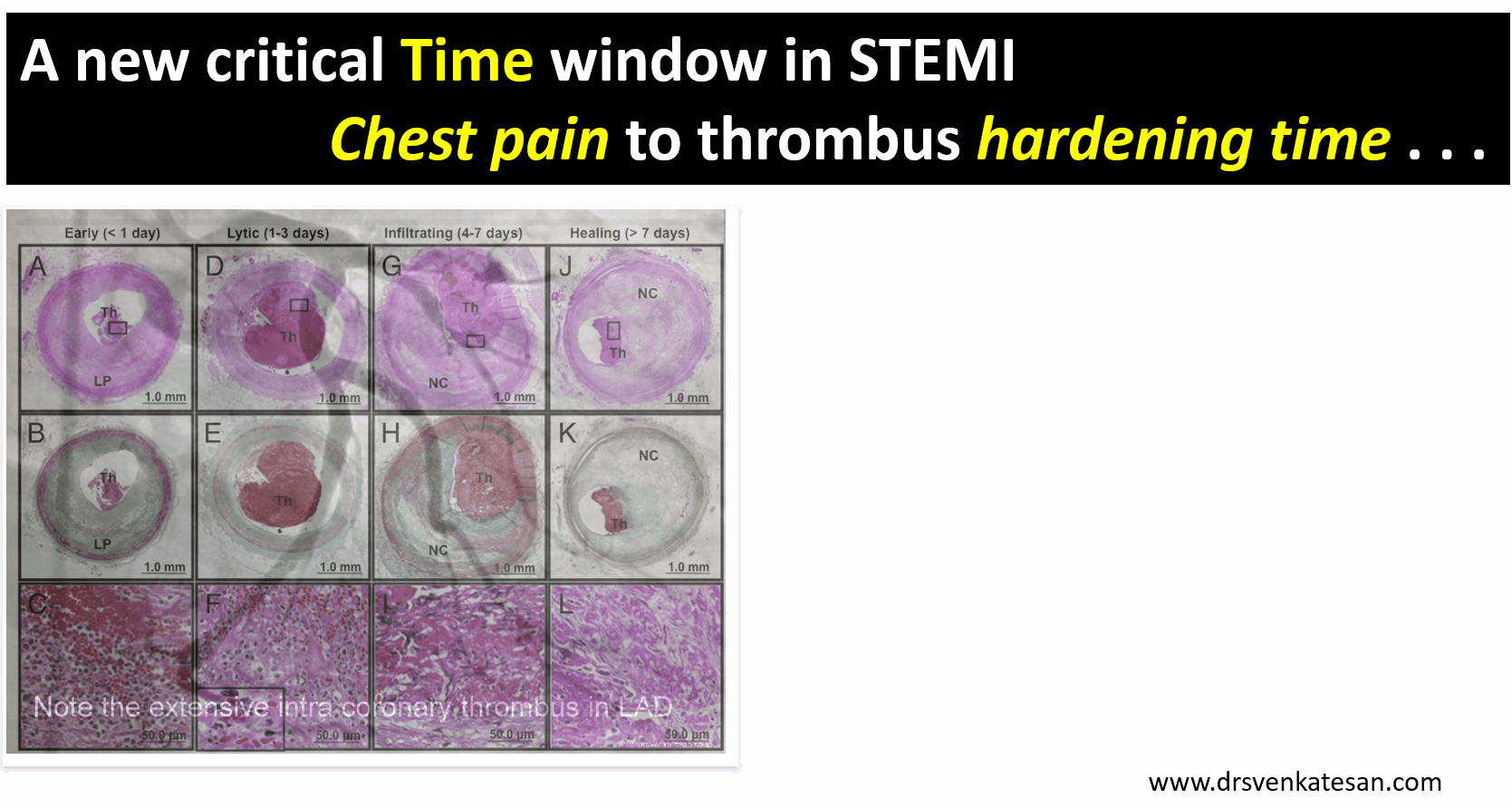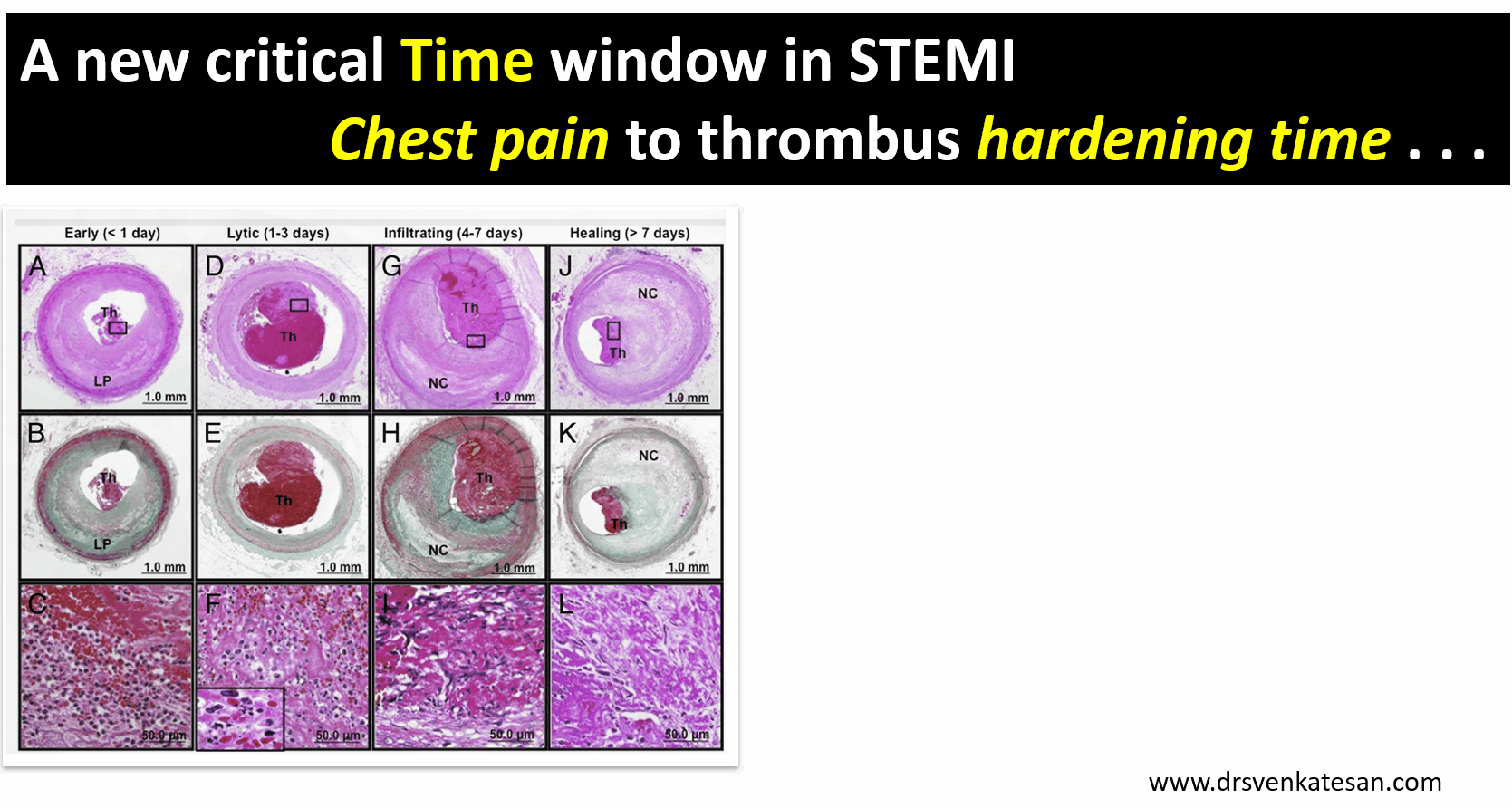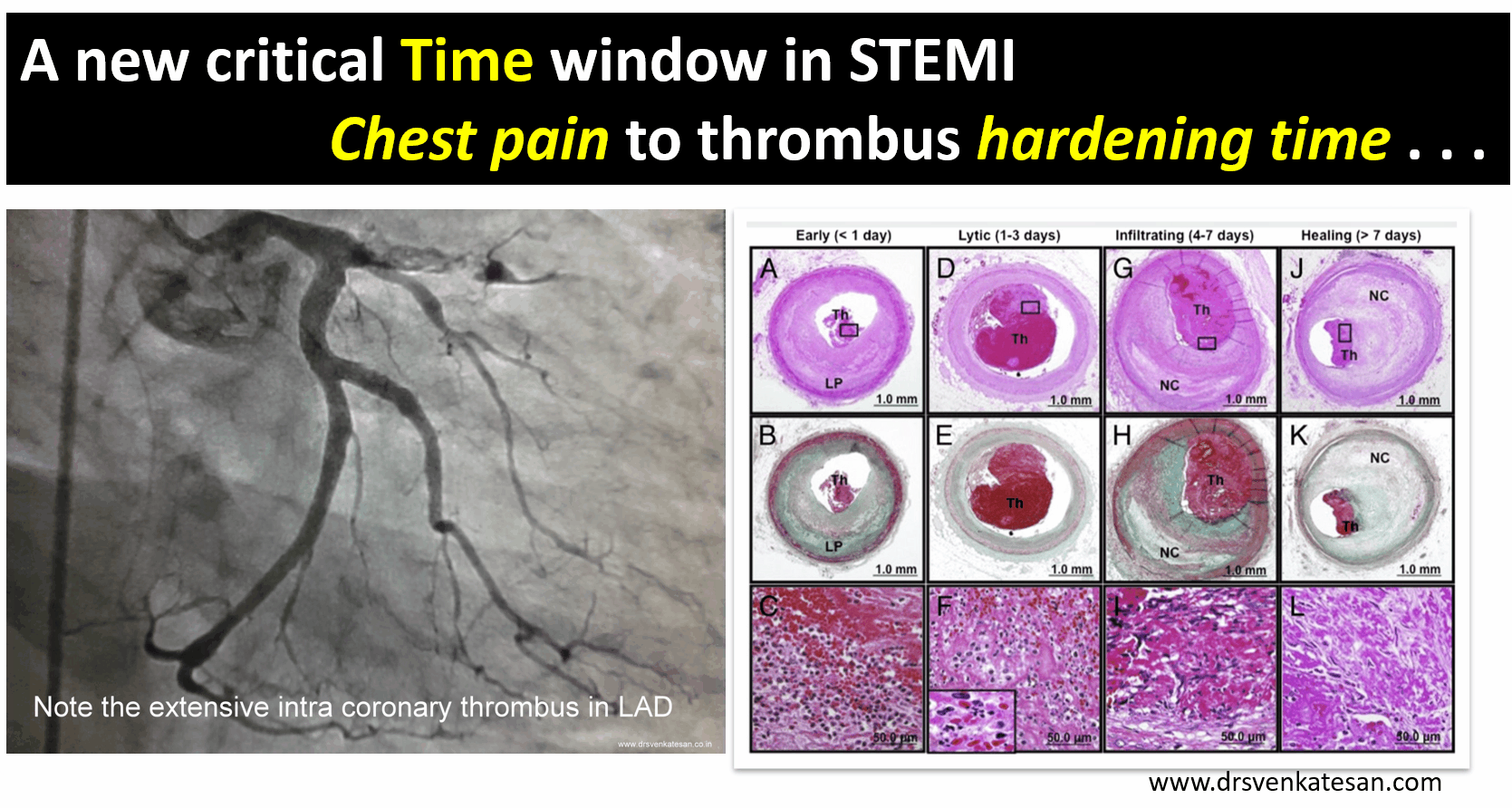
A middle-aged man a Biotech engineer, who is just back from his annual health check, sitting in front of me with a deeply anguished face and said “Doctor my LDL is 130mg, and my diastolic BP is 90 mmHg and fasting sugar is 120 mg .I am very much worried about my future”
Wait , let me go through your file, I said ,
Isn’t a serious Issue doctor?
No, its not ,

But , doctor, I have read about ASCOT, SPRINT and HOPE-3 trials. I guess they tell us to keep the LDL, blood sugar and diastolic BP all these three parameters around 80. Isn’t doctor? He went on to add, that his old fashioned family physician has asked to continue the beta blocker. He said he is also aware of the fact, how JNC has ditched the beta blocker to the graveyard since they don’t do anything to central Aortic pressure. He continued, “Last year my routine coronary calcium score was beyond 300 . Shall I go for a regular coronary angiogram to ensure my SYNTAX score is around zero doctor” ?
I was quite shocked with his academic prelude, and asked him, by any way, he is a physician or a cardiologist?
No doctor, I am purely a non-medical man but follow all health related stuff from wall street medical bulletins. I am a busy man, still, work out regularly. I have important targets, ambitions to fulfill and lot to achieve in life. But, this LDL and BP is really bothering me doctor.
Yes, I got it . . . I understand your anxiety. Don’t get worried about all these biochemistry and hemodynamics. They are just numbers. Some of them will fluctuate to the tunes of your wall street as well.
Really Doctor?
Yes, we are all unlucky, in one sense you know. We are living in a man-made (scientifically) insecure environment. Great men in the past never had to bother with these silly numbers that currently define health. Alexander the great , neither had his Macedonian master health check nor he looked up for his lipid particles, (he was counting his horses Instead) Did Chengiskhan ever knew about his BP ?

If only these men were worried about these fancy number the world history would have been rewritten.
They didn’t even know an organ called the heart that is pumping 5 liters of blood every minute, until Harvey found the circulatory system 1000 years later. Still, they conquered the world. If we take world history millions of men and women have tasted the pinnacle success without really bothering to know their periodic Individual organ function status.
Here is one more story from my country, The Raja Raja Chola the great built this biggest Hindu monument called Brgadheeswara temple in Tanjavur ,Tamil Nadu , India in the year 1010 .

A fictitious query – Who did FFR for Raja Raja Chola (947-1014AD) when he had vague chest pain from suspicious LAD lesion just after his war with Rashtrakuta empire .He went on to Live for 67 years conquering much of India without a single health check and ECG in his life time
It was an unparalleled kingdom of South India where millions of happy men and women who lived a healthy life with absolute faith and trust in their village healers who did the magic with Indigenous leaves, herbs, secret medical formulas based on ancient wisdom.
Longevity with a purpose
The anxiety to live long often keeps our lipids , sugar and blood pressure high . . . and sets a vicious cycle. Today ,this has become a perfect ground for the saviors of health care to trap us in a cartel who are conferred with an almost divine power of defining who is healthy and who is not.
Many times philosophers have felt longevity and the urge to live long, lacks a matching and meaningful purpose. Lack of purpose, as well as extreme obsession with a purpose, are equally dangerous. The purpose of life can never be equated solely on the longevity of our life. Life long fear and anxiety about possible illness and death is not welcome.
Human life span is mystical journey determined by genes as well the environment and its interaction with each other (Epigenetics) It’s destined to face challenges.Substantial of them can be managed without anyone’s help. I will be happy if you don’t ever need the help of cardiologist to get rid of fear and anxiety induced by general health awareness.
Isn’t prevention better than cure Doctor? I came for a possible coronary angiogram . . . but you have really confused me doctor!
No , I am not doing that Intentionally. From your angle its prevention of potential hidden disease. I am talking In a larger perspective, Master health checks many times end up as medical witch hunting. I am bothered about technological contamination that is all too pervasive among the health care system, especially manifesting as new non-existing diseases. (Skewed and tinkered normal curve )
We, the modern men . . . with all six senses intact, tend to make our life miserable with all these digitized biological data and deeply mined medical images from Innocuously good organs. Some times, we seem to more worried about artificial intelligence and least bothered to know the advantages of being naturally ignorant.
Life is not live data that is in motion. Have a good purpose in life, be physically active, think right, eat well, life shall be lived with peace. Please realize many pockets of the world had been more peaceful, healthy, and cultured in the past, than the current glorious and glamorous times. Of course, life expectancy has definitely prolonged with breakthroughs medicine but It’s not clear it has any positive impact in terms of overall global well being.
Please wake up , you are in the middle of patient consult story … Doctor!
Oh yes , thanks. As a parting advice, I sermonized, homo-sapiens are generally programmed to live for about 100 years except in a fraction who have either true incurable disease or those who succumb to a bad fate.
I realized , what should have been a simple prescription for an ARB +Thiazide + Statin and a stress testing , turned out to be an unsolicited compulsive lecture on life’s purpose, and philosophy etc I said sorry to my patient.
He silently got up. His body language was clearly not convincing enough to suggest, he has accepted my confabulations. He left the clinic with a humble thanks probably looking for a more saner physician!
.