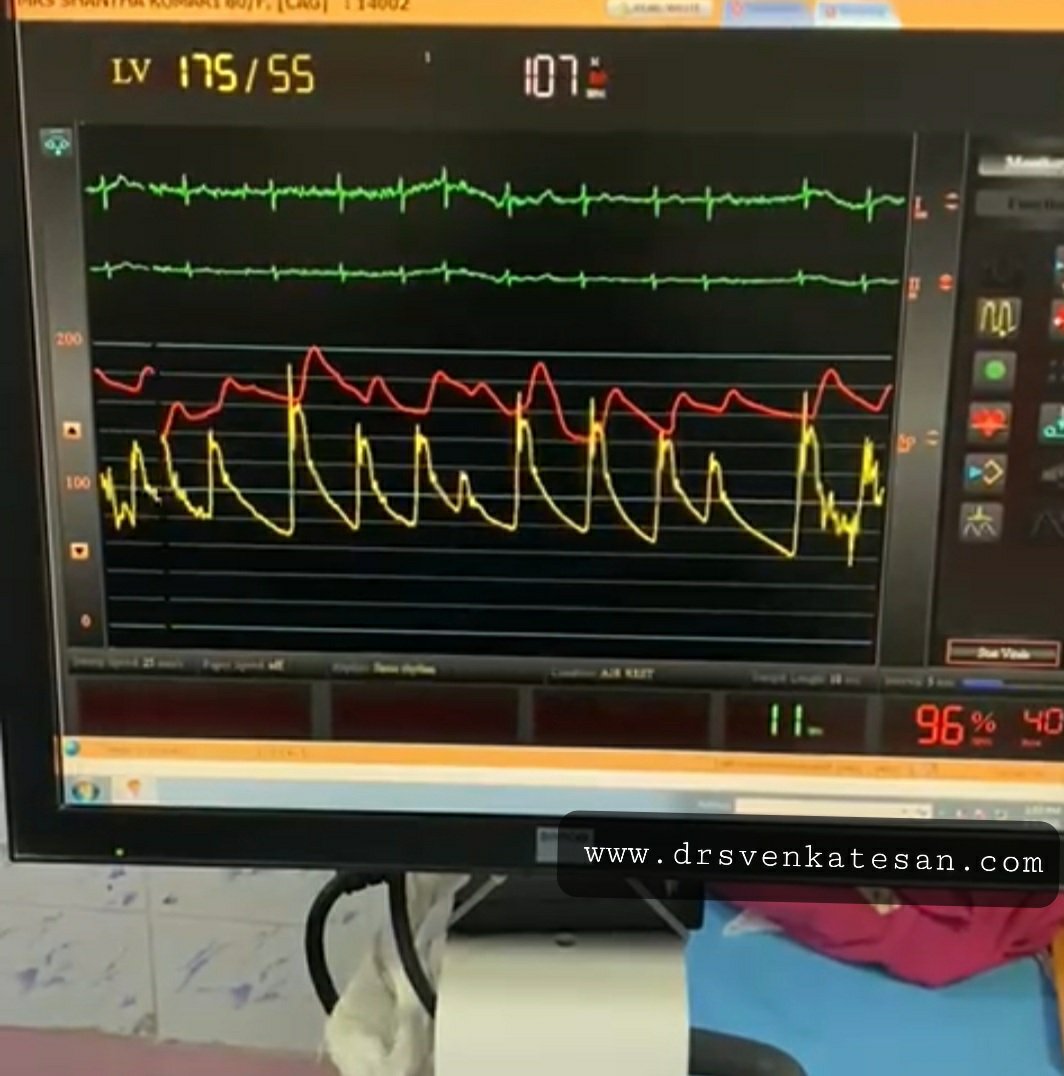
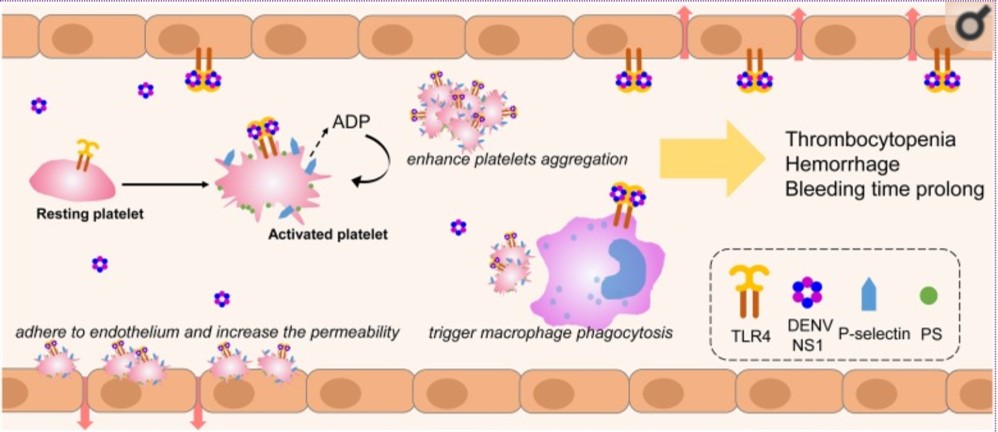
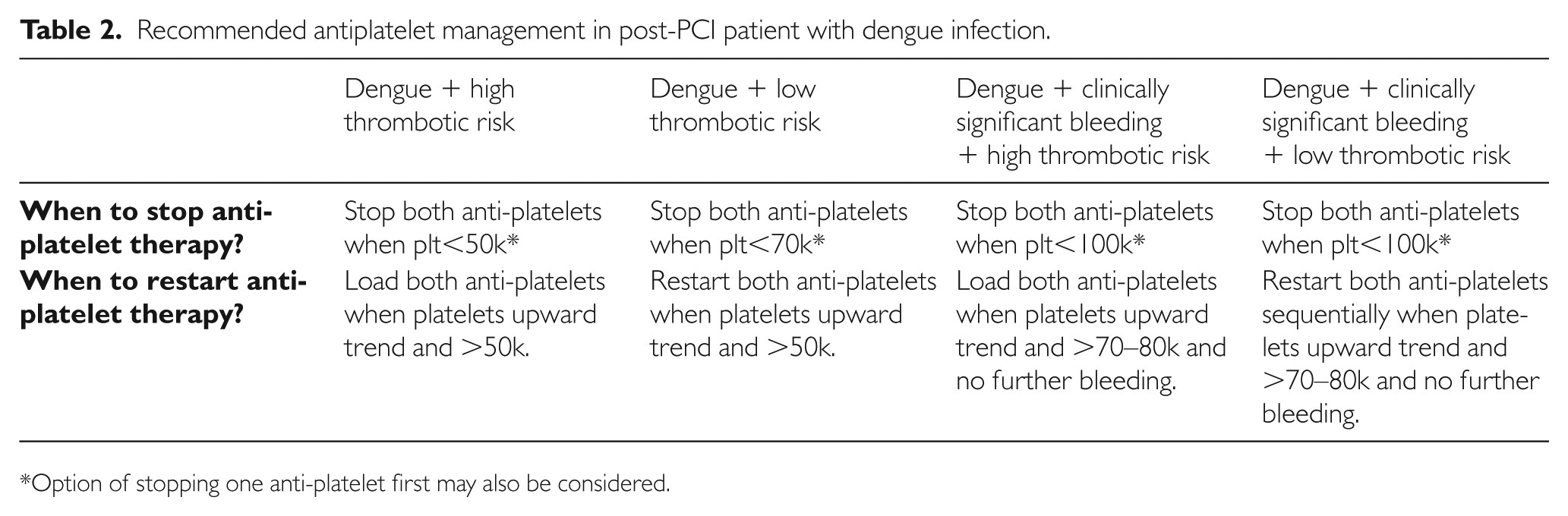
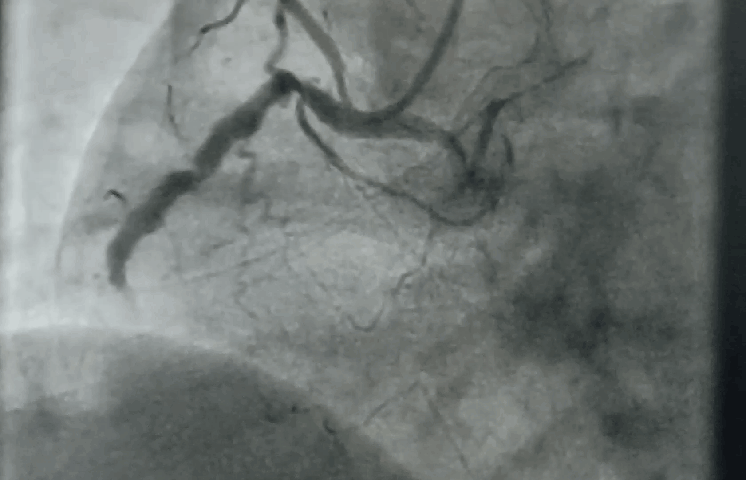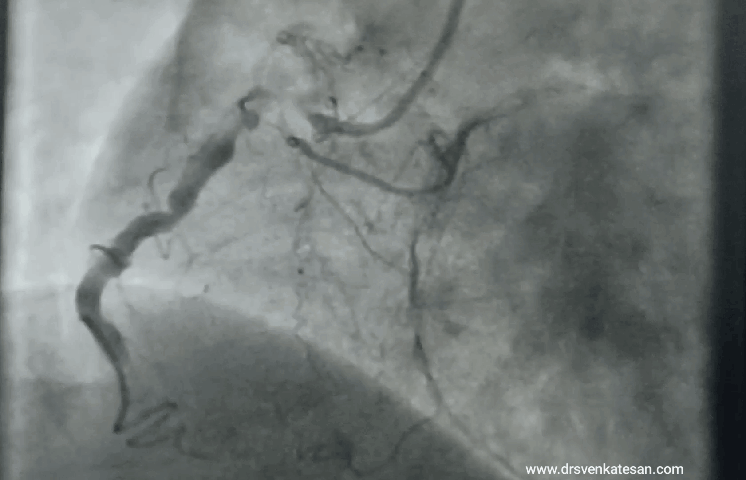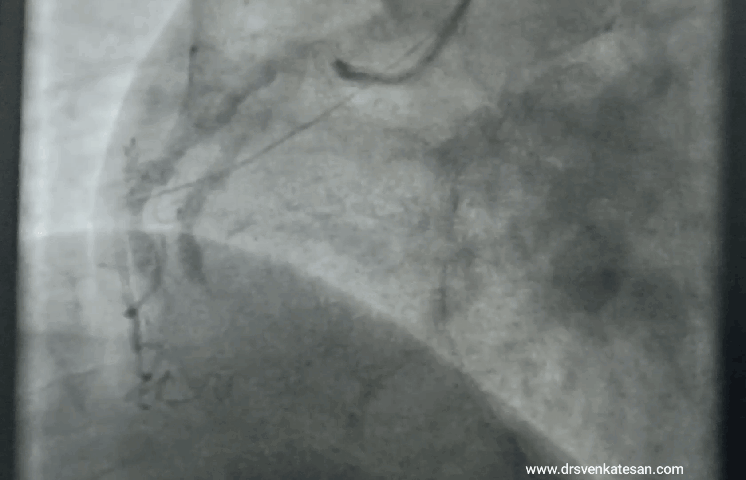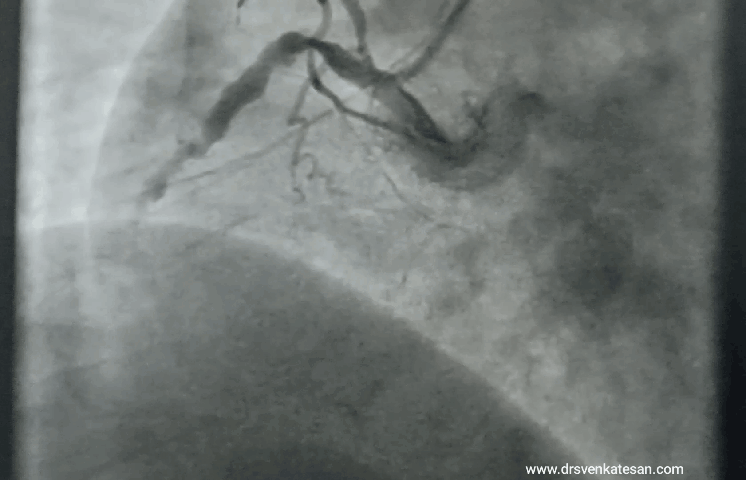
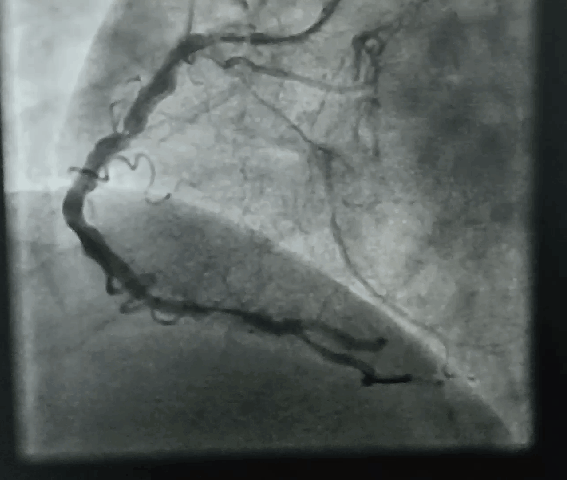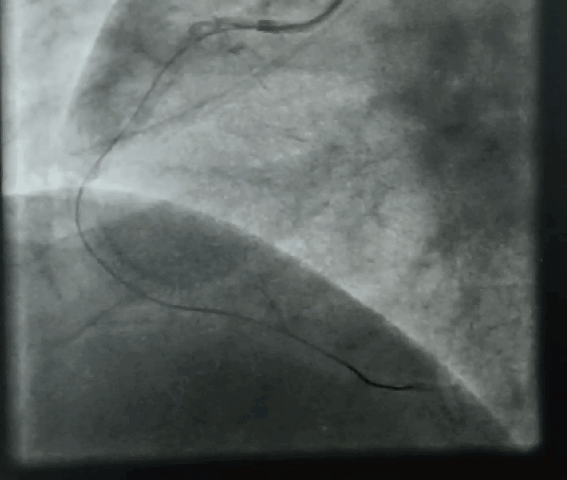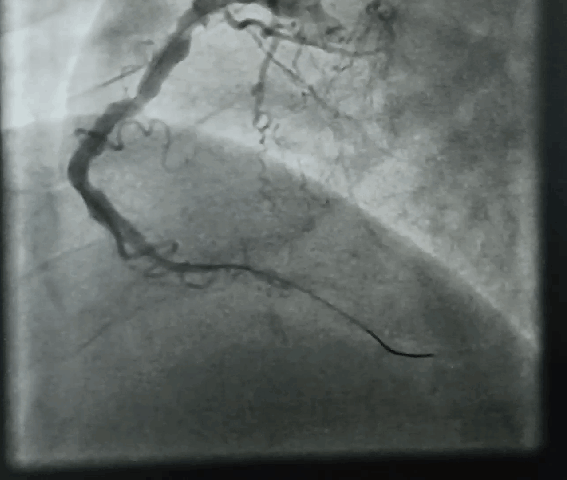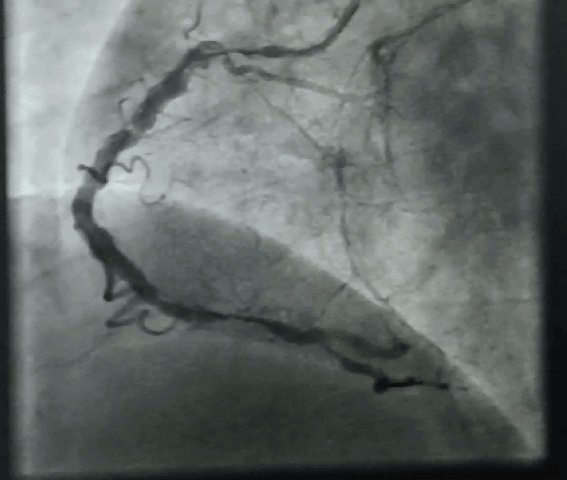
“It was severe double vessel disease & turned out to be a complex angioplasty in LAD ”
Why doctor? what happened?
It was a hard lesion, there was plenty of calcium deposits. It was not clearly visible in the angiogram. I had to do IVUS. Curiously, the calcium was clustered in all the three planes of the vessel ( intima the media and adventitia) and they projected into the lumen blocking the path.
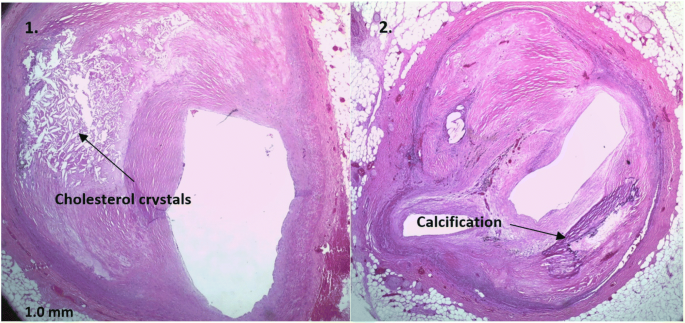

Image collage representation purpose
Thank you, doctor, how did you manage to remove it,?
It was a real struggle. I had to break the calcium shell before deploying the stent. (What we refer to as lesion preparation). I thought Initially I can displace it with high-pressure balloon inflation, I went up to 40 ATM pressure, finally needed a rotational ablation with a burr. ( IVL -Intravascular ultrasound was an option, but may not have helped either because of heavy Intimal load, Angiosculpts, and the cutting balloons (Wolverines) we don’t have)
Was it a risky procedure?
Yes, of course, see the sharp burr rotating 150 thousand resolution per second hugging the coronary artery without damaging it
By the way, why did he accumulate so much calcium inside doctor? Can it be due to his daily calcium supplement doctor?
Oh – my ? that is important new Info. I think that wasn’t noted in the case file. Tell me more, How long did he take it?
I think he is taking it for long years. He is rather obsessed with it, consumes lots of calcium in his diet as well. He was also taking vitamin D as it once went down to 15 ng/ml. He needed to strengthen his bone which was porotic in the Dexa scan. Was that a problem now? But his serum calcium was always normal, Then, from where does, this calcium enter my dad’s coronary artery doctor?
I am sorry I don’t know the answer so far. Now, your dad seems to teach me a lesson. It has to enter from the bloodstream or happen de nova due to degeneration. Calcium along with phosphate is a tightly regulated metabolism.They are closely linked to diet, bone, renal and endocrine glands function. We don’t understand how there can be a no-relationship between blood calcium and coronary arterial calcium. Again plaque calcium and serum calcium may or may not correlate. Understand our knowledge base with which we treat you.
That’s ok doctor. But do you think, should he stop calcium & vitamin D tablets from now on?
Hmm, you keep asking tough questions. I will be a fool if I asked you to continue right!
Please go through this article from Jhon Hopkins and decide ( & MESA study 10 year follow on calcium supplement Ref 2)

Final message
The human body is a wonderful machine. Calcium is a life-sustaining ion present in each of the 75 trillion cells we have. We are programmed to handle all elements except in a fraction when true pathology strikes. Never try to outsmart our natural homeostasis with unnecessary and unsolicited chemicals. Indiscriminate calcium supplements are definitely an unfriendly guest for the coronary artery.
Now, Is it just a coincidence? the new epidemic of coronary microcalcification detected by CT scans(What is your Agatston’s score buddy ?) is matching with a steady increase in per capita consumption of calcium and vitamin D tablets.
Reference
1.Anderson JJB, Kruszka B, Delaney JAC, et al. Calcium intake from diet and supplements and the risk of coronary artery calcification and its progression among older adults: 10-year follow-up of the Multi-Ethnic Study of Atherosclerosis (MESA). J Am Heart Assoc. 2016;5:e003815.
2.Myung SK, Kim HB, Lee YJ, Choi YJ, Oh SW. Calcium Supplements and Risk of Cardiovascular Disease: A Meta-Analysis of Clinical Trials. Nutrients. 2021 Jan 26;13(2):368. doi: 10.3390/nu13020368. PMID: 33530332; PMCID: PMC7910980
For advanced readers
1. Does calcium stabilise a lesion or destabilize a lesion?
It does both .
2. Is CT calcium score correlate with serum calcium?
Yes, it does.Ref : Sanghoon Shin, European Heart Journal, Volume 33, Issue 22, November 2012, Pages 2873–2881, https://doi.org/10.1093/eurheartj/ehs152
3. What are the various types of coronary calcium? Which is a more tricky lesion?
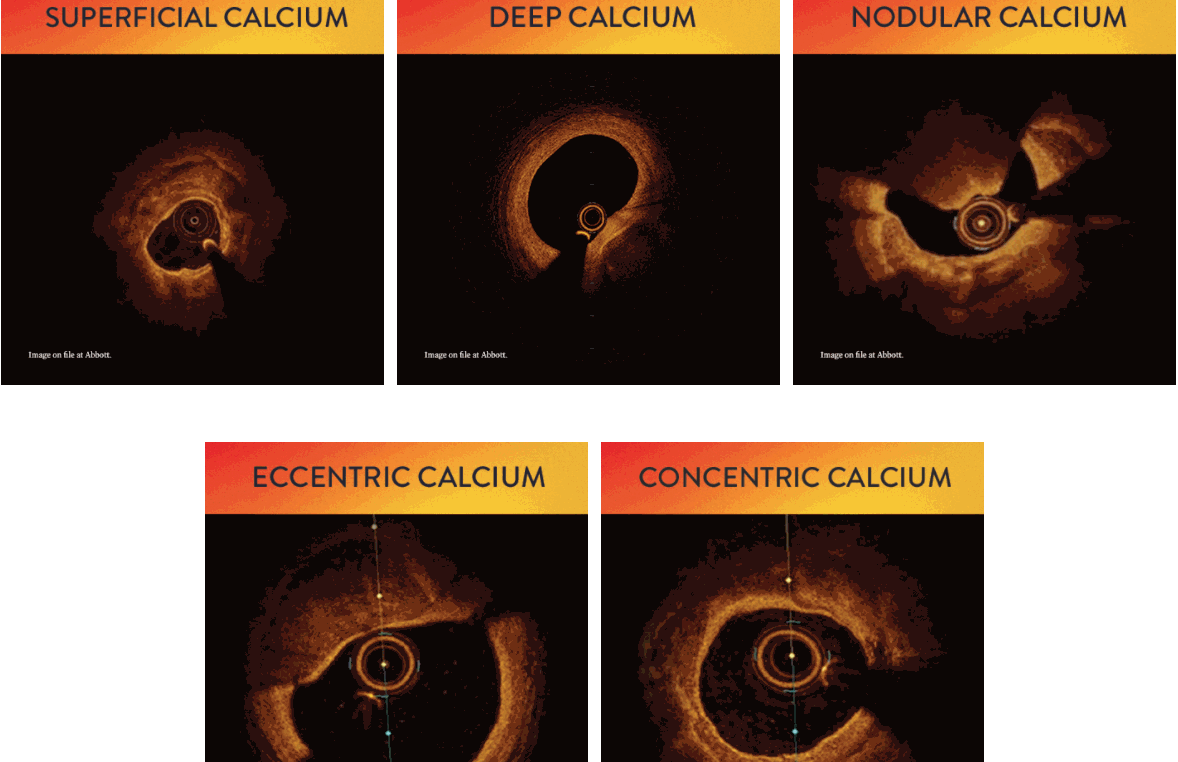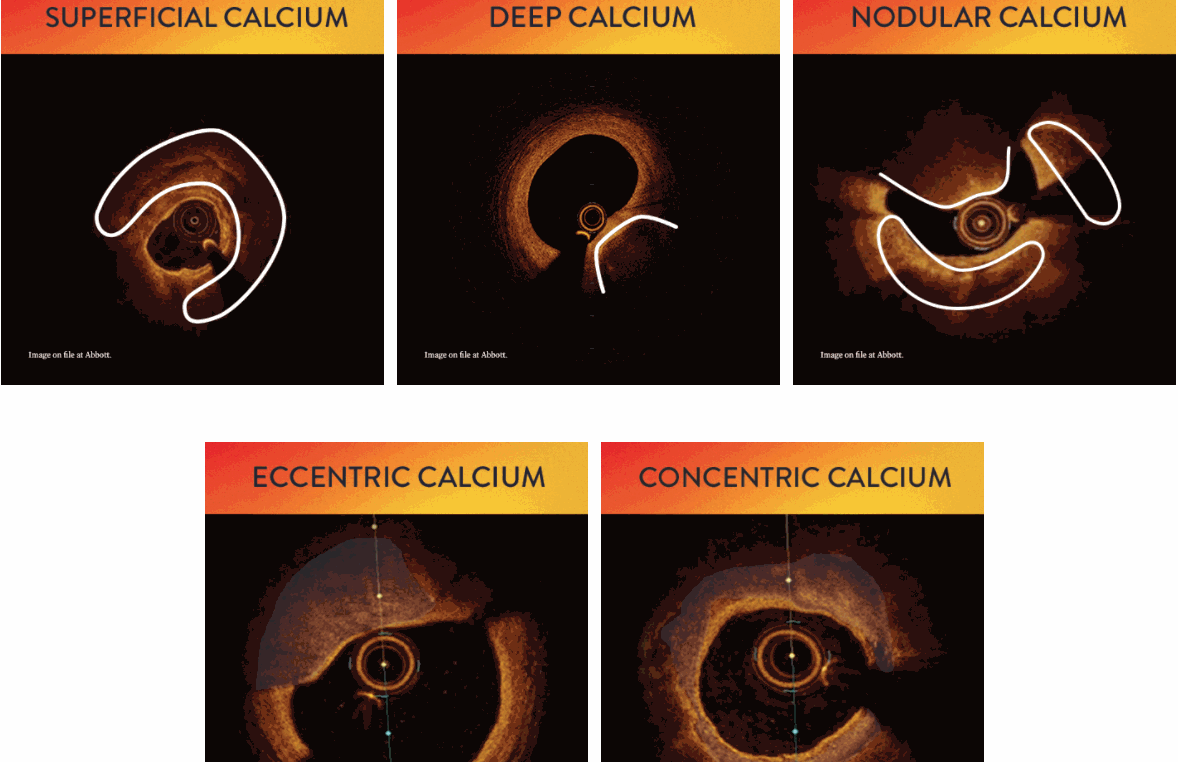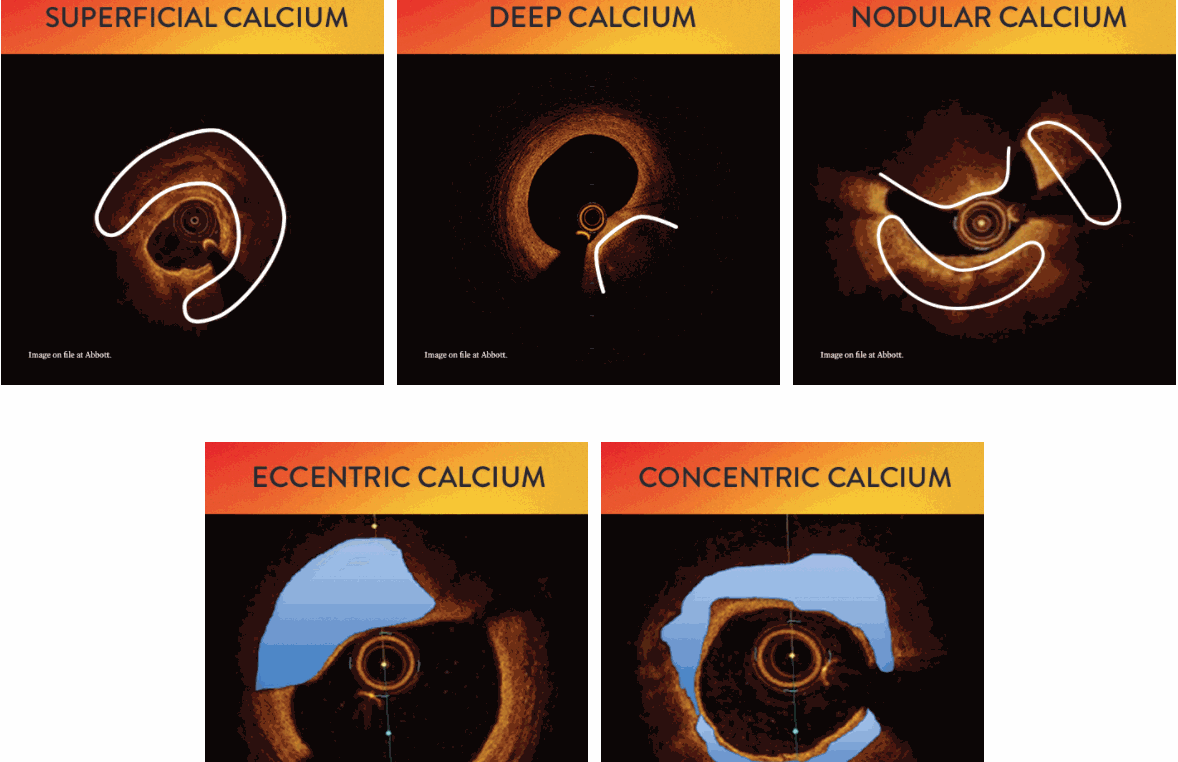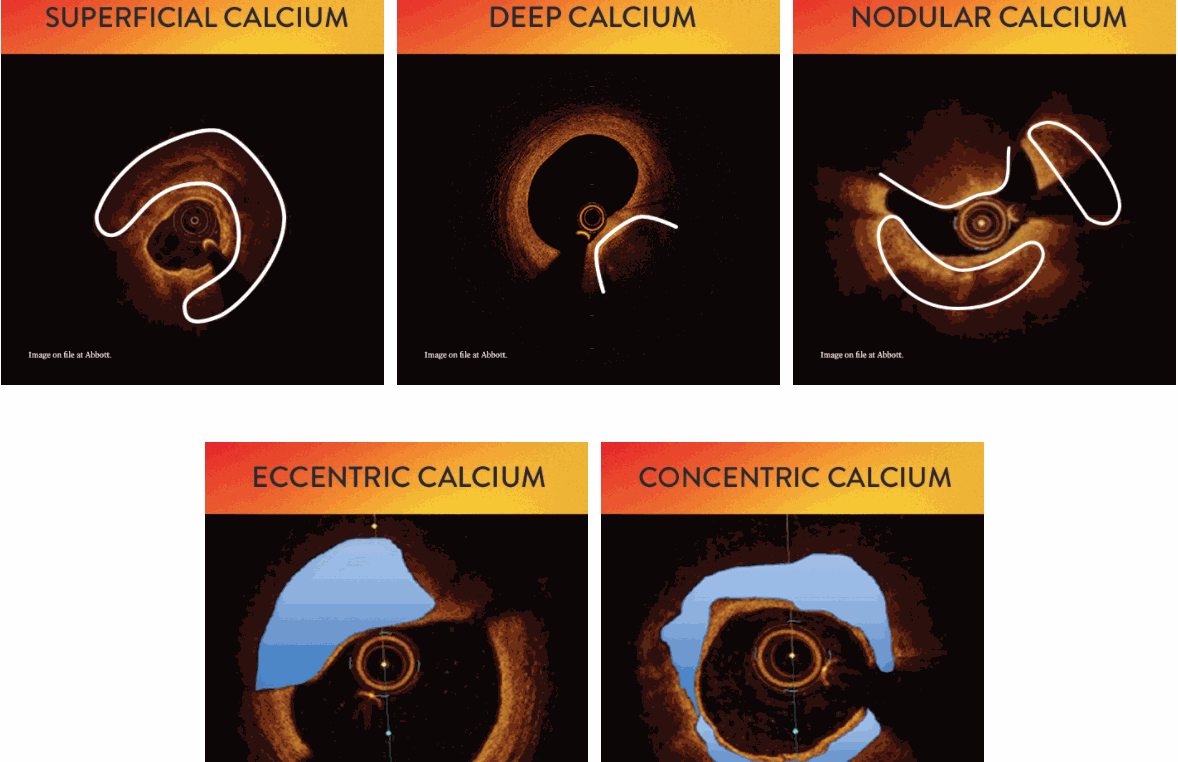
Image courtesy -Abbot A calcific nodule is the tricky one, as it can be tiny still result in invisible stent malapposition inviting future problems.