Left atrium is the posterior most chamber of the heart. It is almost a mid line structure. The normal size of left atrium is about 4 / 4 cm. Normal left atrial volume is 46ml in men and 38 ml in women .(Atrial volume in a normal adult population by two-dimensional echocardiography Y Wang, Chest, Vol 86, 595-601.) Left atrium is not an easy chamber to identify in the X ray chest as it does not form the cardiac border.( Except a small circumference of left atrial appendage.(LAA)
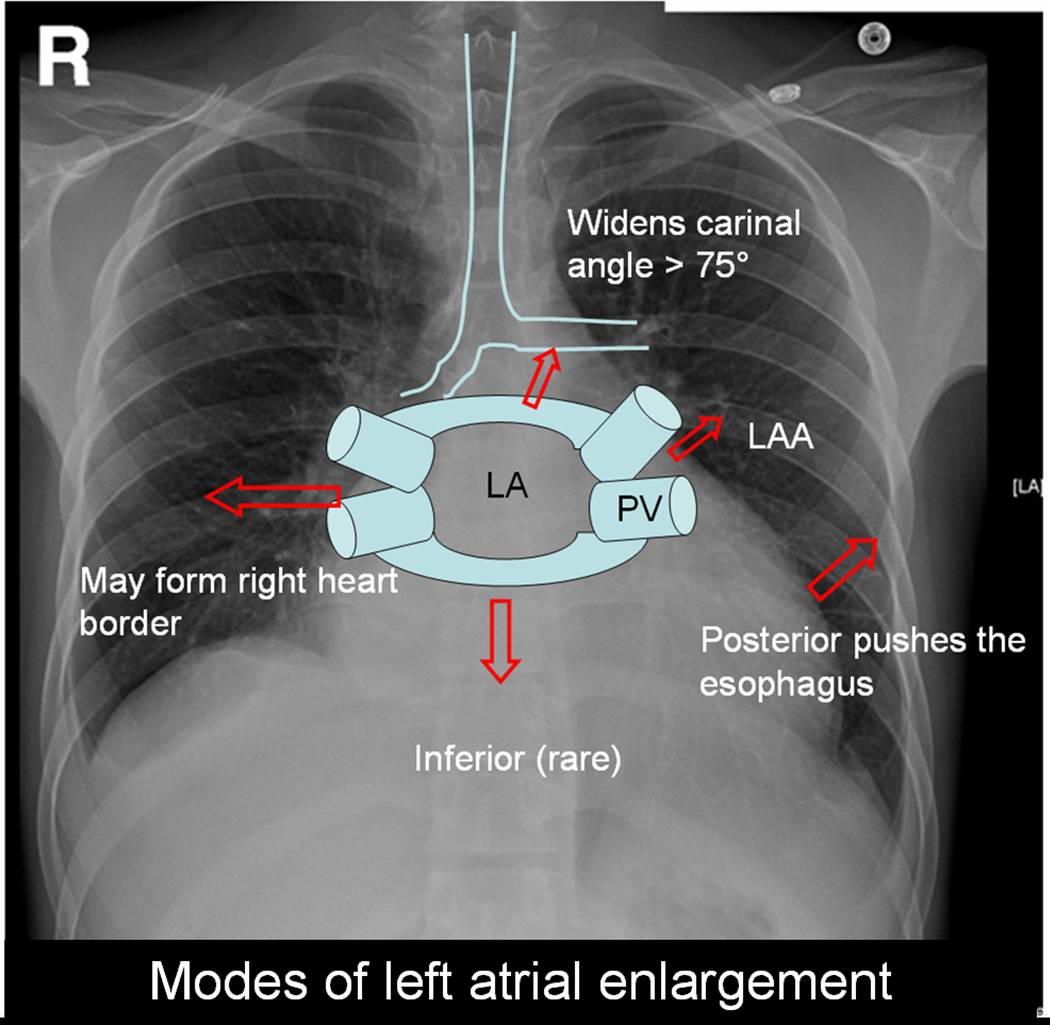
Left atrium can enlarge in multiple directions.Generally it dilates in the path of least resistance.

- It is believed left atrial appendage enlargement occur early . LAA enlargemnet seen as a fullness beneath the pulmonary artery shadow. It may be the earliest finding of LAE in X ray. ( This may appear as straight left heart border , as in classical mitral stenosis where MPA is also enlarged). The LAA enlargement is not necessarily in in proportion with LAE.
- LA could also enlarge posteriorly by pushing the esophagus towards the spine.This is visible only in barium swallow.
- Then LA can enlarge either to left or right ( Usually towards right) and reach the right heart border or over shoot it and form the right heart border by itself.This occurs very late in the course.
- The other direction LA goes on to enlarge is superiorly. When LA enlarges superiorly it hits on the left main bronchus and lifts it.This is measured by the widened subcarinal angle which is normally less than 75 degrees.
- LA can enlarge anteriorly sometimes , but it is resisted by right ventricle but rarely right ventricle yields to the LA push and produce a left parasternal lift which could be mistaken for RV enlargement.
- Inferior enlargement can not happen in a significant way as it is limited by the AV groove and strong fibrous skeleton.
With the advent of echocardiography X ray assessment of LA is redundant .(Academic value and in fellows training programs).The upper limit of normal LA size is around 4.5cm.
LA enlargement is commonly seen in
- Rheumatic mitral stenosis, regurgitation. Gross enlargement up to 10 cms are common.
- Hypertensive heart disese.
- Cardiomyopathy, especially restrictive where both atria enlarge.
In all these conditions if atrial fibrillation occurs LA size increases further.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}